
第16讲 Immunotherapy
HEADS UP!
Antibodies manufactured in the lab are being used to treat disease. Some therapeutic antibodies bind to cytokines or growth factors and prevent these proteins from signaling. Others tag cells for destruction. Therapeutic antibodies also can be used to block the action of checkpoint proteins. This can give T cells a better chance to fight cancer. And T cells removed from a patient can be modified and infused back into that patient as a cancer therapy.
INTRODUCTION
Currently, there is great interest in using "immunotherapies" to treat human diseases such as autoimmunity and cancer. In this lecture, I will discuss some examples of the approaches that are being tried, sometimes with great success. What you will notice is that all of these immunotherapies are based on an understanding of how the immune system normally functions to protect us from infections.
IMMUNOTHERAPY USING MONOCLONAL ANTIBODIES
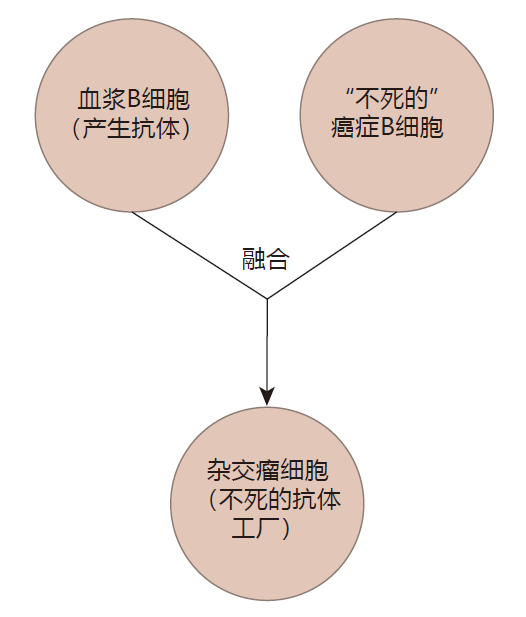
Antibodies have properties that make them extremely useful to the immune system: They can bind tightly to a specific target, and can direct the immune system to destroy that target. These same properties also could make antibodies useful in treating disease. But there is a problem: Plasma B cells produce a ton of antibodies, but plasma B cells have a very short lifetime – usually only a few days. So for antibodies to have practical value as "drugs," a procedure would need to be devised that would extend the life of plasma cells. Two scientists named George Köhler and Cesar Milstein recognized that many cancerous blood cells are "immortal" and can be grown in the lab almost indefinitely. They hypothesized that if they could somehow fuse a cancerous B cell that didn't make any antibodies with a B cell that was making the antibody they wished to mass produce, they might be able to create a hybrid cell – a hybridoma. Ideally, this hybrid would combine the best qualities of both of its "parents": A hybridoma could make large quantities of the desired antibody, and, because it could be grown indefinitely in the lab, it could be used as an antibody "factory." Although this sounds a bit like science fiction, their idea worked! Indeed, this discovery was so important that Köhler and Milstein were awarded the Nobel Prize. Because the hybridoma technology results in a clone of immortal cells that produces only one kind of antibody, the antibodies that are made are called monoclonal antibodies.
Currently, about half of all drugs in clinical trials are monoclonal antibodies, and a number of monoclonal antibodies have already been approved for immunotherapy by the Food and Drug Administration (FDA) in the United States. The common term (e.g., in a TV ad) for a therapeutic monoclonal antibody is a "biologic."
Using monoclonal antibodies to treat autoimmune disease
Some therapeutic monoclonal antibodies are designed to bind to specific proteins and keep them from functioning. The inflammation associated with rheumatoid arthritis is caused mainly by tumor necrosis factor (TNF), a cytokine produced by macrophages which infiltrate the joints under the direction of self-reactive helper T cells.
Monoclonal antibodies (e.g., Humira (adalimumab)) can block the action of TNF, either by binding to TNF or to its receptor. TNF blockers can be very effective in decreasing the severity of arthritis symptoms, and these monoclonal antibodies are now the world's most popular class of drugs, with annual sales that exceed US$25 billion. These antibodies are administered by injection under the skin, usually every two weeks. Although TNF blockers work well, it is important to keep in mind that TNF is a cytokine which is an important part of the immune defense. Consequently, inhibiting its action leaves patients susceptible to infections. I'm sure you have heard the long list of "disclaimers" when these drugs are advertised on TV!
Plaque psoriasis is another autoimmune disease that is being treated with monoclonal antibodies. The "bad actor" in this disease is the cytokine IL-17, which can cause skin cells (keratinocytes) to proliferate when they should not. This errant proliferation results in the patches (plaques) of thickened and scaling skin that characterize the disease. Monoclonal antibodies (e.g., Cosentyx (secukinumab)) that block the interaction between IL-17 and its receptor on keratinocytes are quite effective in treating moderate-to-severe plaque psoriasis. However, one of the normal functions of IL-17 is to help defend against fungal infections (e.g., Candida albicans). Consequently, patients who are treated with IL-17 blockers have an increased susceptibility to yeast infections.
Monoclonal antibodies also are being used to eliminate immune system cells that are responsible for causing disease. Campath-1H (alemtuzumab) is a monoclonal antibody that binds to CD52, an antigen that is abundant on the surface of B cells, T cells, and monocytes – but not on the surface of other cell types. When Campath-1H binds to CD52, it can trigger destruction of its target cell either by fixing complement (and overwhelming the cell's anti-complement defenses) or by antibody-dependent cellular cytotoxicity (ADCC), in which the monoclonal antibody identifies the target and a phagocyte does the killing. Importantly, although this monoclonal antibody can deplete B and T cells, it does not destroy the blood stem cells that produce these lymphocytes. As a result, after the immunotherapy has been discontinued, new B and T cells can be made to replace the ones that have been destroyed. Campath-1H is currently used to treat multiple sclerosis, an autoimmune disease mediated by self-reactive T cells.
The monoclonal antibodies pioneered by Köhler and Milstein were made by fusing two mouse cells – so they were mouse antibodies. Consequently, these antibodies could be seen as "foreign" by the human immune system and destroyed, limiting the time that they would survive in the body of a patient. To circumvent this potential problem, genetic engineering can be used to replace most or all of the foreign DNA sequence that encodes the antibody molecules with the corresponding human sequence. As a result, the patient's immune system will be tolerant of these "humanized" monoclonal antibodies. Campath-1H was the first humanized antibody to be approved by the FDA.
Using monoclonal antibodies to treat cancer
Monoclonal antibodies are being used to treat several types of cancer. Rituximab is a monoclonal antibody that binds to a protein called CD20 on the surface of B cells, and marks these cells for destruction by antibody-dependent cellular cytotoxicity. This was the first monoclonal antibody approved by the FDA for the treatment of cancer, and it has been used very successfully to treat non- Hodgkin lymphoma – a blood cell cancer which arises when B cells suffer mutations that block their maturation process.
CD20 is expressed on the surface of immature B cells (e.g., non-Hodgkin lymphoma cells), but it is not found on the surface of blood stem cells which function to "restock" the blood system. Moreover, CD20 is not expressed on B cells that have matured to the antibody-producing stage. The idea here is that rituximab will bind to the CD20-expressing lymphoma cells and tag them for destruction, yet will spare blood stem cells and long-lived plasma B cells which, as a consequence of an earlier infection or vaccination, are producing protective antibodies.
Roughly 25% of patients with metastatic breast cancer have tumors which produce unusually large amounts of a growth factor receptor called Her2. When this surface receptor is ligated by growth factor proteins, it causes these cancer cells to proliferate. The monoclonal antibody Herceptin (trastuzumab) can bind to the Her2 receptor, "covering" it, and preventing it from receiving "grow" signals. As a result, for the subset of patients whose breast cancers overproduce Her2, this immunotherapy can increase survival time by slowing the growth of metastases.
In Lecture 8, we discussed how two "checkpoint" proteins, CTLA-4 and PD-1, appear on the surface of activated T cells to keep them from becoming over- exuberant.
T cells which undergo repeated rounds of activation and proliferation express increasing amounts of CTLA-4 on their surface. This checkpoint protein competes with the activation receptor CD28 for binding to B7 (expressed on activated dendritic cells), and makes it harder for T cells to be reactivated in secondary lymphoid organs. This can limit the ability of tumor-specific T cells to build up their numbers to the point where they might be numerous enough to destroy a tumor.
Ligation of the checkpoint protein PD-1 does not interfere with activation. Rather, ligation of PD-1 suppresses the effector function of T cells (e.g., the ability to kill their target cells) and their ability to proliferate. Indeed, the main purpose for PD-1 expression appears to be to dampen the immune response and minimize the "collateral damage" to tissues which might result if T cells continued to function after an infection had been cleared.
Tumor cells frequently express PD-L1, the ligand for PD-1, and other cells in the tumor environment can be induced to express PD-L1 in response to cytokines such as IFN-γ, which result from the inflammation associated with a tumor. By expressing or inducing the expression of PD-L1, solid tumors can "protect themselves" by creating a local environment that is hostile to the T cells which otherwise might destroy them.
Immunologists reasoned that if cancer patients do have T cells which can target their tumors, these cells might be held in check by either or both of these checkpoint proteins. If so, it might be possible to "reinvigorate" the anti-tumor immune response by treating patients with monoclonal antibodies that would block the interaction between the checkpoint proteins on T cells and their ligands. One of the first checkpoint inhibitors to reach the market was a monoclonal antibody called ipilimumab that can bind to CTLA-4 on the surface of T cells and prevent this checkpoint protein from "soaking up" the limited number of B7 proteins on APCs. This type of checkpoint blockade has been most effective in treating metastatic melanoma, and has extended the life of some patients. However, one of CTLA-4's normal functions is to provide protection against autoimmunity by making it harder to reactivate self-reactive T cells that are chronically stimulated by plentiful self antigens. Consequently, monoclonal antibody blockade of CTLA-4 can result in serious side effects such as colitis and liver inflammation – conditions that are commonly associated with autoimmune disorders.
More recently, monoclonal antibodies have been created that can bind either to PD-1 on T cells or to PD-L1 and block the interaction of these two proteins. PD-1 blockade seems to cause less serious autoimmune-type side effects than does CTLA-4 blockade, and PD-1 blockers have been used to treat about a dozen different cancers with response rates ranging from 15% to 90%. Hodgkin lymphoma, advanced melanoma, and lung cancer are among the cancer types which have been successfully treated. Even for bladder cancer, where the response rate is only about 15%, and where untreated patients typically survive for less than one year, treatment with monoclonal antibodies that block the PD-1/PD-L1 interaction has extended the life of some patients for more than three years. Probably the most famous patient to have received PD-1 blockade therapy is Jimmy Carter. In 2015, President Carter was diagnosed with a malignant melanoma that had metastasized to his brain and liver. The prediction was that he would live only a matter of months. He was treated with a combination of radiation, chemotherapy, and a PD-1 blocker – and is still alive three years later.
It is important to note that checkpoint blockade only works as a cancer treatment if a patient's immune system is already making anti-tumor T cells whose effectiveness is limited either because there are too few of them or because they do not function well. Although checkpoint blockade has been useful in treating some cancers, tumor-specific T cells are not found in the majority of human tumors, indicating that most cancer cells do not activate the adaptive immune system. In patients who do have tumor-specific T cells, it has been found that most of these T cells have receptors that bind to neoantigens – "new" antigens which cancer cells make as a result of mutations in the DNA that encodes normal cellular proteins. As a consequence of such mutations, neoantigens are essentially "foreign" antigens – antigens to which CTLs are not tolerant.
In general, immunotherapy with antibodies that block the PD-1/PD-L1 interaction work best if the patient's tumor cells express high levels of PD-L1. Hodgkin lymphoma cells, for example, have a genetic mutation that causes them to overexpress PD-L1, and the response rate for treatment of this cancer with PD-1 blockade approaches 90%. Unfortunately, Hodgkin lymphoma is the exception. Checkpoint blockade generally can extend the life of only about 20% of people with certain other cancers. Moreover, there is currently no good way to predict who the "lucky" 20% might be.
Well-established tumors that contain cells with many genetic mutations have a greater likelihood of producing neoantigens that can be recognized by T cells. However, such tumors also have a higher probability of including "escape" variants – cancer cells in which the neoantigen can no longer be presented or recognized. Consequently, although the positive responses to checkpoint immunotherapy can last for years, most patients' tumors do not disappear completely, and many tumors that regress or are stable as a result of checkpoint blockade begin to grow again after a relatively short time. Finally, checkpoint inhibitors are administered by infusion once every two or three weeks in a hospital, and these treatments are expensive: This monoclonal antibody treatment currently costs more than US$100,000 per year per patient.
The functions of CTLA-4 and PD-1 are non-redundant.
CTLA-4 acts mainly in the secondary lymphoid organs to prevent T cell activation. In contrast, PD-1 usually functions at the site of the cancer as a negative regulator of the anti-cancer response. Consequently, clinical trials are now underway to test whether blocking both of the these checkpoints is more effective than just blocking one or the other, and to determine how toxic this combination therapy might be.
IMMUNOTHERAPY USING T CELLS
T cells also can be used to treat disease. In some cases, this involves assisting "natural" T cells that just need help getting the job done. In other cases, T cell immunotherapy uses T cells that have been modified by genetic engineering to make them "better, faster, stronger."
Cancer immunotherapy using adoptive cell transfer
When surgeons removed tumors from patients with cancer (e.g., melanomas), they often found that the cancerous tissue had been "infiltrated" by T cells – cells which they named "tumor infiltrating lymphocytes" or TILs. When these cells were examined, it was discovered that some of the TILs had receptors which could recognize antigens expressed by the cancer cells. This finding suggested that the immune system was trying to deal with the cancer, but that there might just be too few tumor-specific T cells to do the job.
To test this idea, immunologists devised the following procedure: They divided the cells recovered from a patient's tumor, and cultured these cells independently in the presence of IL-2 to cause the tumor infiltrating lymphocytes to proliferate. Next, each culture was tested to identify the one which contained T cells that had the highest activity against the cells of the tumor. This "winning" culture was forced to proliferate further to produce about 100 billion tumor-specific T cells. Finally, these "living drugs" were infused back into the patient to treat his cancer.
This procedure, usually called adoptive cell transfer (ACT), has resulted in the cessation of tumor growth and even tumor eradication in some melanoma patients. In one trial, the tumors of 20 of 93 patients regressed completely, and 19 of these individuals experienced no recurrences when tested five to ten years after they had been treated – indicating that they probably were cured. Nevertheless, for the majority of patients, such "victories" are not long-lasting, and their cancers ultimately progress.
So far, only melanomas have yielded TILs that are useful in ACT.
Adoptive cell transfer has several advantages. This immunotherapy relies on the expansion of naturally occurring, tumor-specific T cells, and it is not necessary to know which antigen(s) is recognized by the TILs that are isolated. Also, most TILs target neoantigens. As a result, even when a very large number of TILs is administered, these treatments generally don't cause autoimmunity. On the other hand, because TILs usually recognize mutated proteins that are unique to each patient, ACT is outrageously expensive.
Cancer immunotherapy using engineered T cells
The goal of adoptive cell transfer is to greatly increase the number of a patient's natural tumor-specific lymphocytes so that they are numerous enough to win a battle the patient's immune system is already fighting. However, this type of immunotherapy fails if tumor-specific T cells do not exist or cannot be isolated. In addition, TILs only can destroy cancer cells whose MHC molecules are able to present the tumor antigen which the TILs recognize – and cancer cells are notorious for mutating to prevent antigen presentation. Moreover, tumors are genetically heterogeneous, so some tumor cells may express the TILs' target antigen, whereas other cells in the tumor may not. To circumvent these potential problems, immunologists are exploring ways to use genetic engineering to "upgrade" a patient's T cells, and to use these manipulated T cells to treat that patient's cancer.
Although a number of different approaches are being used to engineer T cells to fight cancer, the therapy that has given the best results so far is called "chimeric" antigen receptor (CAR) T cell therapy. The name comes from a Greek mythological creature which had the head of a lion, the body of a goat, and the tail of a dragon. The concept behind CAR T cell therapy is to use genetic engineering to modify a patient's T cells so that they produce an "artificial" T cell receptor. Like the mythological creature, this synthetic TCR usually has three parts: First there is a recognition domain that is displayed on the surface of the engineered T cell, and which can bind to the desired surface antigen on the target cancer cell. This recognition domain is usually a single heavy/light chain binding region "borrowed" from an antibody molecule that recognizes the target antigen. This extracellular recognition domain is connected to a second protein inside the cell that contains the T cell's CD3ζ protein, which can signal that the target receptor has been engaged. And to provide the necessary co-stimulatory signal, the CD3ζ protein is joined to the signaling domain of a co-stimulatory molecule like CD28. The idea here is that the recognition domain of the CAR T cell will identify the target, the CD3ζ protein will send the TCR engaged signal, and the CD28 domain will provide the required co-stimulation – all in one chimeric protein. Pretty amazing!
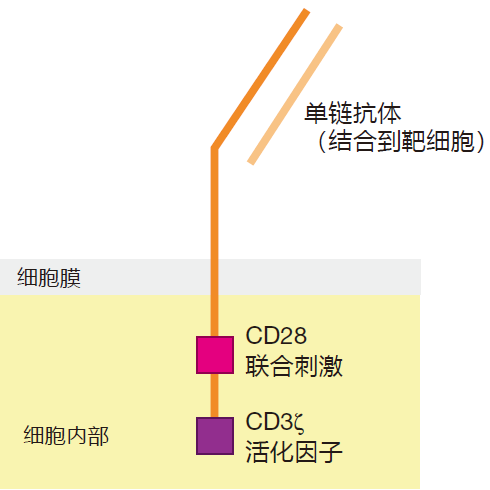
So CAR T cells are "designer" T cells with enhanced capabilities. The genes that encode the chimeric protein usually are inserted into the genome of a lentivirus (e.g., a version of HIV-1, modified so that it is non-pathogenic), and this virus is then used to infect T cells that have been harvested from the patient's blood. This "carrier" lentivirus can incorporate its genetic information, including the engineered CAR construct, into the genome of the infected T cell so that when the cell proliferates, all of its progeny will express the chimeric receptor protein. The virus-infected T cells are then forced to proliferate in the lab to build up their numbers, and are infused back into the patient. As you can imagine, this type of genetic engineering ain't easy. CAR T cell immunotherapy, in which T cells are "repurposed" or "redirected" to destroy cancer cells, is the result of many thousands of hours of research over a period of more than 20 years. Interestingly, the use of CAR T cells was the first gene transfer therapy to be approved by the FDA.
Although CAR T cells are being developed with recognition domains that can bind to various antigens on the surface of cancer cells, the target which has been most successful in the clinic is the protein CD19. This protein is part of the B cell's co-receptor, which binds to opsonized antigens, and whose normal function is to make it easier for antigens that are decorated with complement proteins to activate B cells. Importantly, CD19 is expressed on the surface of most leukemias and lymphomas, and CD19 CAR T cell therapy has been used successfully to treat two types of B cell malignancies: acute lymphoblastic leukemia and non-Hodgkin lymphoma. The goal of CD19 CAR T cell therapy is to destroy all the B cells in a patient's body which express CD19. This protein appears on the surface of B cells early in their development and continues to be expressed until B cells are about to become plasma cells. So the result of depleting B cells that express CD19 is that B cells which have already become plasma cells are spared, but B cells that have not matured to the plasma cell stage (including the cancerous B cells) are destroyed. Of course, not having B cells which can protect against new invaders is not a good thing, because it can put the patient at risk for life-threatening infections, so patients typically are given gamma globulin to help them fight infections. A recent trial used CD19 CAR immunotherapy to treat 45 children and young adults with acute lymphoblastic leukemia. About 90% of them went into remission as a result of the treatment, but about half of these relapsed within a year.
CAR T cells are engineered to recognize antigens expressed on the surface of their target cells (e.g., CD19).
Consequently there is no requirement that the receptors of CAR T cells recognize antigens presented by MHC molecules. This avoids the problem of cancer cells "hiding" by mutating to disrupt the antigen presentation machinery. Nevertheless, many of the patients who
relapse after CD19 CAR therapy do so because mutations in the gene for CD19 make the cancer cells "invisible" to the engineered receptor. So escape mutations are still a problem with CAR T cell immunotherapy. Moreover, part of the "magic" of antigen presentation is that CTLs are able to look at peptides that normally are found inside the target cell. The receptors of CAR T cells can only look at surface proteins (e.g., CD19), so the number of potential targets for CAR T cell therapy is limited.
One must also be very careful which target one chooses for CAR T cell therapy. T cells with natural TCRs have undergone testing for tolerance of self antigens, but CAR targeting domains have not received this training. Consequently, cells chosen for CAR T cell elimination must be cells that are not essential for human health. And CAR T cell therapy is not without side effects.
Roughly one-third of the patients in early trials suffered from serious neurological problems, including hallucinations, delirium, and seizures. Immunologists are working hard to "fi ne tune" CAR T immunotherapy to try to deal with these undesirable side effects.
So far, most of the successes with CAR T cell therapy have been with blood cell cancers such as leukemias and lymphomas. Success with solid tumors has been limited. One reason is that blood cell cancers are easier to target than solid tumors because we can live without certain types of blood cells – at least temporarily.
In contrast, most of the "easy" targets on the surface of solid tumors also are found on cells that are essential for life, and targeting these shared antigens could cause life-threatening autoimmunity. This is unfortunate because solid tumors are responsible for about 90% of all cancer fatalities.
At present, CAR T cell immunotherapy is very complicated and unpleasant for patients, and is usually used as a "last resort" for people with an otherwise desperate prognosis. Novartis' CAR T cell treatment for children with end-stage leukemia was the first therapy using engineered T cells to reach the market. This immunotherapy can result in long-lasting remissions and even cures.
However, because CAR T cells must be engineered for each individual patient, this highly personalized therapy is costly: about US$500,000 per patient.
Other approaches which employ the weapons of the immune system to treat cancer are in various stages of testing. We can all hope that these experiments will be successful – because, as it stands now, about one out of every three of us will get cancer. But please remember one thing: It is estimated that 20–40% of all cancers can be prevented by living a healthy lifestyle.
REVIEW
Hybridomas are made in the laboratory by fusing a B cell that produces a desired antibody and a cancerous B cell that can live forever. The monoclonal antibodies produced by these "antibody factories" can be used to treat autoimmune disease and cancer. Some monoclonal antibodies block the interaction between cytokines or growth factors and their receptors. Other monoclonal antibodies recognize antigens on the surface of cells (e.g., cancerous B cells) and mark these cells for destruction. Monoclonal antibodies that block the binding of the checkpoint proteins CTLA-4 and PD-1 to their ligands can "reinvigorate" T cells that are tumor-specific.
T cells also can be manipulated and used to treat cancer. Adoptive cell transfer uses naturally occurring, tumor-specific T cells (TILs) that have been isolated from individual patients, and grown in culture to increase their numbers. CAR T cells are "designer" T cells with enhanced capabilities. They are made by using genetic engineering to equip T cells with artificial T cell receptors that can recognize cancer cells without the requirement for antigen presentation by MHC molecules.