
1.5 生物衰老学家如何研究衰老:系统生物学
在本节中,我们将讨论系统生物学,这是一种用于准确预测生物学结果的数学建模技术。一个系统可以定义为一组相互连接的事物或组成一个复杂整体的一部分。美国国立卫生研究院的Ron Germain将系统生物学定义为一种建模过程,力求“将工程学、数学、物理学和计算机科学的原理与广泛的实验数据相结合,发展对生物现象的定量和深入的概念性理解,从而预测和准确模拟复杂的生物行为。”
系统生物学主要关注复杂系统中各个部分之间的关系或联系。这些关系可以通过数学和计算机科学领域的图形理论进行研究,这种理论专门研究图形的性质和应用(图1.20)。当然,计算方法超出了本文的范围。本节的目的是简要介绍系统生物学以及如何使用系统生物学预测衰老的生物学结果。我们重点介绍系统生物学的背景、定义和理论。本节末尾提供了图形分析在生物衰老学中的应用。 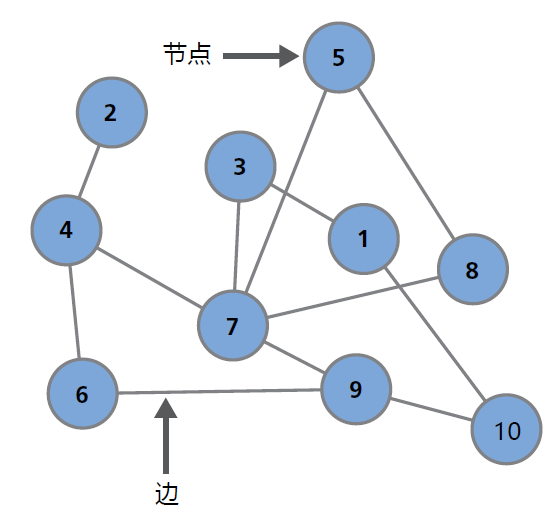
图1.20 称之为网络的数学图形。网络中有数字编号的对象称为节点或顶点,连接节点的线称为边,到任何一个节点的连接数称为度。
系统生物学将有助于将生物学转变为预测科学
系统生物学代表了几种新的定量方法之一,这些方法将生物科学从主要的观测科学转变为定量科学。定量(数学)方法提供了在复杂系统中可证明、准确预测的能力——这种能力到目前为止一直是物理科学的专属领域。许多人认为,这些新的定量方法将会建立准确预测和模拟复杂生物行为所需的两个关键要素:(1)普遍的生物规律,相互作用要素之间真实、绝对和不变的关系;(2)生物常数,每次测量结果都是一样的。虽然我们找到能够预测方程的第一个真实和普遍的生物学定律和常数可能还需要几十年的时间,但系统生物学已经坚定地证明,在生物科学中,定量方法和预测的力量是可能的。
那么,为什么系统生物学方法会有利于生物衰老学呢?首先,随年龄增长而来的功能丧失反映了内部和环境因素或变量之间的相互作用;衰老是一个极其复杂的问题,其交互成分几乎是无限的(图1.21)。第二,世界上没有两个人一辈子生活在完全相同的环境中,因此每个人的年龄都会不同。任何关于未来功能丧失的预测都必须通过分别考虑每个个体的内部和环境变量之间的相互作用来进行(见第2章中关于精确医学的讨论)。考虑到影响衰老的因素几乎是无限的,只有计算机的计算能力才能单独得出预测未来功能损失的方程式或算法(假设已经建立了定律和常数)。一旦建立了预测某个功能丧失的方程式或算法,我们就可以设计出干预预防或延迟功能丧失的方法。因此,我们就可能实现更长更健康的寿命。
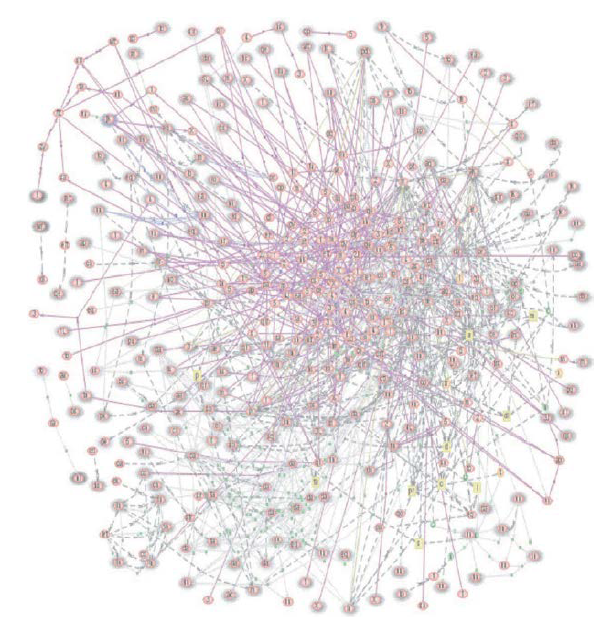
图1.21 基因/蛋白质网络图。本图显示了线虫中与衰老/寿命相关的基因之间的联系。有202个基因(节点)产生4000多种蛋白质。(摘自Witten TM,Bonchev D.2007.Chem Biodivers 4[11]:2639-2655.经John Wiley and Sons许可。)
科学的还原方法(Reductive method)已成为生物学研究的特点
生物学和医学的研究在很大程度上以自上而下或还原论的方法为特点。还原法是基于这样一个概念,即通过生物系统的各个部分可以更好地理解生物系统。还原法从对生物现象的广泛观察开始。然后,进行多个实验,以便将系统分解或简化为各个部分,直到对初始观测结果得出最可能的解释。例如,雀鸟喙大小的变化最终让达尔文推断所有生命都是从一个点进化而来的。他推断,通过生殖传播的一种可改变的因素是造成生命形式变化的原因。在接下来的100年里进行了数千次实验,将可能的可修改因子减少为一个DNA。达尔文对所有生命的共同因素的研究一直在减少,从一开始对复杂系统进行观察,最后到DNA分子结构的研究(图1.22)。 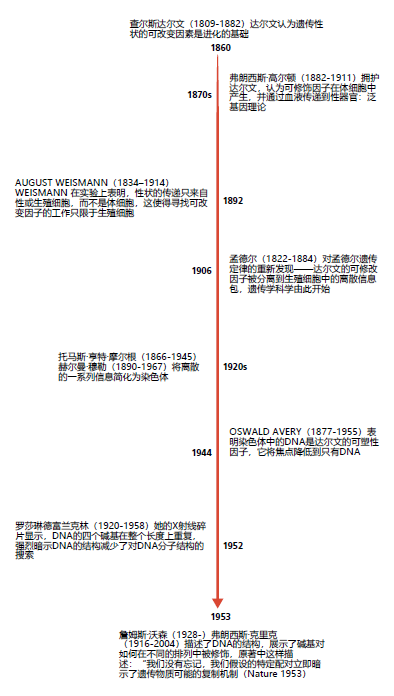
图1.22 描述还原法如何应用于DNA结构发现的大致里程碑。
还原法应用于生物学和医学的重要性怎么强调也不为过。它归功于上个世纪在生物学和疾病治疗方面取得的显著发现。尽管如此,即使是还原论得出的最详细的结果也只是具有高度概率的观察结果,通常被称为强推断(strong inference)。还原论通过归纳推理(inductive reasoning)的方式推断机制,此过程通过观察得到结论。由于强推断仅仅是通过一个正确率很高的结果来归纳推理结论,因此结论缺乏准确性。应用于还原论观察的可证明的定量方法创造了一门精确的预测科学。
系统生物学和还原论共同增加知识和改进预测
系统生物学和其他定量方法不会取代还原科学。相反,系统生物学提供了一种方法,通过该方法,从还原法中得出的结果可用于模拟生物过程的数学模型(图1.23)。为了进一步说明观测科学和数学如何一起用于预测结果以及精确数学预测对科学的重要性,我们使用牛顿引力定律的一个高度简化的例子(图1.24)。我们没有使用生物科学的例子,因为系统生物学还没有发现一个普遍接受的方程来预测生物学结果。 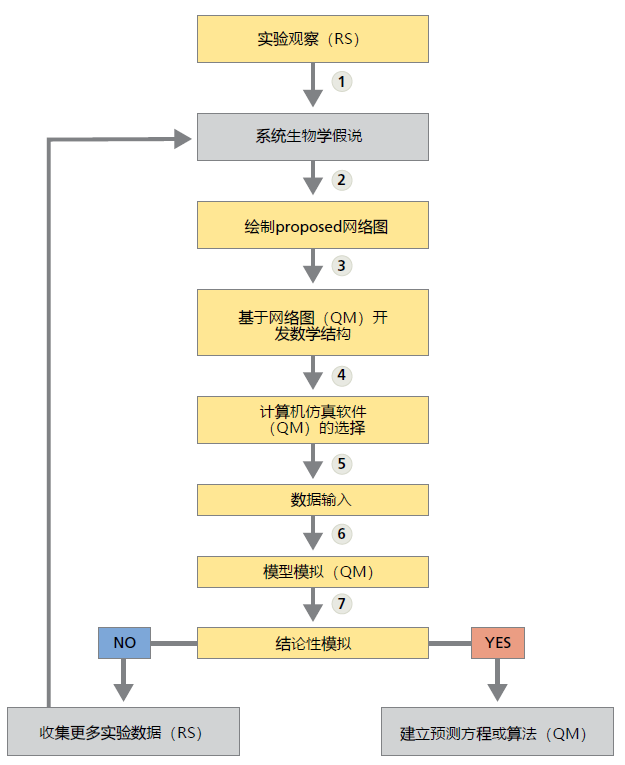
图1.23 系统生物学中建立预测方程和算法的步骤。此步骤还展示了主要使用还原科学方法(RS)或定量方法(QM)的步骤。(1)实验观察提供了建立假设的信息(2);(3)拟定(proposed)网络图提供了系统生物特性的概要,有时称为系统边界,系统边界的估计建立必要的基本数学方法;(4)数学方法将确定运行模型模拟所需的软件类型;(5)将从数据库(见下文)检索到的适当数据加载到软件中;(6)对模型进行了测试。(7)如果模拟测试提供的结果与实验观察结果一致,则可以开始建立预测方程和/或算法的过程。如果模拟结果与实验观察结果不一致,则需要更多的实验数据和新的假设。

图1.24 牛顿引力定律。这个方程描述了在预测方程时观测科学和定量方法之间的关系。质量(m1和m2)和距离(r)通过观测方法确定。引力常数是通过数学推导得出的。这条定律是重力估算的模型,直到1798年亨利·卡文迪什(1731-1810)根据经验确定了重力常数的值。
在牛顿的引力方程之前,一些天文学家观察到地球是圆的,围绕着很大的太阳旋转,并且有一个静止的轨道。这表明,一定有某种无法解释的力量将人类固定在地球上,并将地球保持在稳定的轨道上。伽利略的实验观察表明,距离和重量(后来显示为质量)是这种无法解释的力的关键因素。牛顿利用伽利略的观测,认识到无论质量大小,这个未知的力都是恒定的,并影响所有有质量的物体。牛顿能够通过微积分“模拟”伽利略的观测,并为重力所涉及的恒力创建一个数值。从这里开始,对牛顿来说,利用观测科学、质量和距离得出的数据,生成一个能够精确预测作用在任何物体上的重力的方程——牛顿引力定律——是一件相对简单的事情。
根据引力定律所做的预测使得太空计划成为现实,但这还需要300年。也就是说,如果没有牛顿引力定律的预测能力,火箭离开地球引力所需的速度是不可能的。以此类推,延迟或预防特定的时间依赖性功能丧失的干预措施将仍然具有挑战性,直到开发出准确的预测方程。
还原论可以预测简单生物系统中的集成性质(emergent properties);复杂系统需要定量方法
系统的行为或结果称为集成性质。集成性质是系统的一个属性,系统的各个成员没有该属性。例如,DNA的四个碱基腺嘌呤、胞嘧啶、胸腺嘧啶和鸟嘌呤的排列方式使得它们具有携带遗传信息的作用。这四个碱基中没有一个能单独遗传。遗传是DNA四个碱基之间相互作用的集成性质。
在一个相对简单的系统中,强推理通常提供足够的信息来充分预测集成性质。传染性病毒性疾病单核血球增多症(mono)可以从其部分预测(诊断),这部分通过还原科学描述的部分。血液中存在的Epstein-Barr病毒准确地预测了单核细胞增多症的集成性质。单核细胞增多症代表了一个相对简单的系统,在这个系统中,一个单一的部分,即Epstein-Barr病毒,可以预测单核细胞的集成性质。我们现在知道,生物学和医学中的许多系统,包括衰老,都比传染病复杂得多。这些复杂的系统包含数千个相互作用的部分,这些部分可能导致多个集成性质。通过还原科学得出的强有力的推论速度太慢,并且缺乏预测复杂系统的集成性质所需的精度(考虑一个只有五个相互作用部分的小系统将有55个或3 125个可能的相互作用)。
衰老和长寿是一个复杂的系统,系统生物学将会帮助我们研究衰老和和长寿,以预测集成性质。在第5章中,您了解到,通过实验观察强推断,一个单一的基因调控着秀丽隐杆线虫(C.elegans)的寿命,这是一种小型线虫(蠕虫)。这一发现表明,长寿也许是一个小而简单的系统。随后的实验表明,名为age1的基因仅仅只是200多个参与长寿调控的基因之一(见图1.21);长寿是一个复杂的系统。此外,一个与age1相同的遗传途径的基因(称作daf16)被发现与调节生长有关。这意味着一个具有长寿集成性质的复杂系统与另一个具有增长集成性质的复杂系统相关联。因为400个基因似乎参与调节生长的集成性质,所以长寿与生长遗传途径的联系表明600个独立基因之间可能存在相互作用。长寿调控的复杂性并不止于此。daf16基因似乎通过被称为雷帕霉素靶标(TOR)的生化网络与免疫系统相连(该途径中的基因数量尚未完全阐明)。把这些放在一起会产生非常复杂的连接排列。只有计算机的计算能力才能产生方程,预测复杂系统中的集成性质,如衰老和寿命。
现代系统生物学和“生物组学(omics)”科学始于人类基因组测序
您已经了解到,系统生物学和还原科学一起工作,以增强对集成性质的预测。还原科学为系统生物学提供了必要的数据,用以发展相互作用部分之间的数学关系,并试图预测结果。这意味着系统生物学只有在还原科学产生准确且足够大的数据时,才能有效地作为预测工具。在人类基因组测序之前,还没有技术能够产生足够数量的精确数据,使系统生物学成为一种有效的预测工具。随着20世纪90年代人类基因组计划的实施,这一切都发生了变化。快速准确地分析DNA中的碱基对序列的技术得到了发展。正如人类基因组计划主任弗朗西斯·柯林斯(Francis Collins)所说,“开发更好、更便宜、更快的DNA处理技术……是启动大规模人类基因组测序的关键里程碑。”一旦这些技术被开发出来,就产生了足够的数据来构建有意义的数学算法,进而帮助准确预测人类基因组的序列。现代系统生物学就诞生了。
虽然人类基因组计划描述了DNA中的碱基对序列,但许多基因和调控序列的功能仍然未知。换言之,系统的部分(也就是基因)在它们的集成性质(即蛋白质)之前就已经产生了。确定每个基因编码的蛋白质的需要使得一个新的研究领域的建立,即基因组学,基因组学是研究整个基因组功能和结构的。
很快科学家发现,DNA高速测序和基因组学领域的工程策略可以应用于生物学的其他领域。因此,高吞吐量(high-throughput)技术的发展有助于确定RNA(跨组学、跨组学)、蛋白质(蛋白质组学、原组学)、表观遗传学标签(表观基因组学、表观基因组学)和细胞代谢物(代谢组学、代谢组学)的整体研究。反过来,从这些新的“生物组学”研究领域产生的数据将可供使用系统生物学方法预测复杂系统中出现的集成性质的研究人员使用。
这些新的高速分析技术可以在相对较短的时间内生成大量数据。当然,如果系统生物学家不容易获得这些数据,那么它们将毫无用处。幸运的是,科学界已经非正式同意建立包含生物组学科学产生的信息的自由开放数据库。最初,联邦政府为主要涉及基因组学的数据库提供物理存储空间。近年来,政府和研究机构维护的所有生物组学科学的开放存取数据库激增。因此,当系统生物学家需要系统某一部分的信息时,他或她可以访问数千个免费数据库,以满足特定需求(图1.25)。 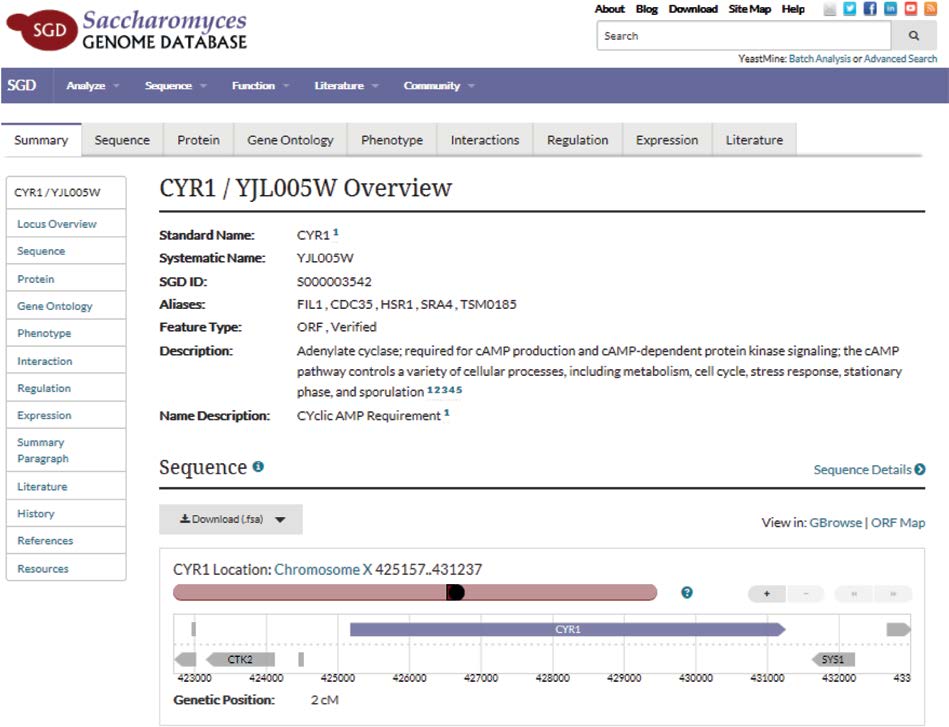
图1.25 酿酒酵母基因CYR1搜索的结果。有关该基因的信息包括此摘要和左侧框中列出的项目。酵母菌基因组数据库(SGD)由斯坦福大学维护。[经Stacia R.Engel博士许可使用](SGD项目。http://www.yeastgenome.org/locus/S000003542/overview#; 2-23-2016.)
生物网络提供了评估系统内相互作用的方法
我们强调,系统生物学和其他定量方法最终将实现精确的预测方程和生物过程的模拟。为了实现这一目标,必须首先确定系统关键部分之间的交互。系统生物学通过对生物网络的分析来确定系统各组成部分之间的相互作用。生物网络是单个部分的集合,单个部分可以形成亚单位或网络,这些亚单位或网络依次连接到构成整体的其他亚单位(图1.26)。网络连接可以可视化,网络特性可以通过计算机软件进行评估。在这里,我们讨论了一个来自衰老生物学的例子,作为对网络分析的初步介绍,酿酒酵母(S.cerevisiae)中的寿命调节网络。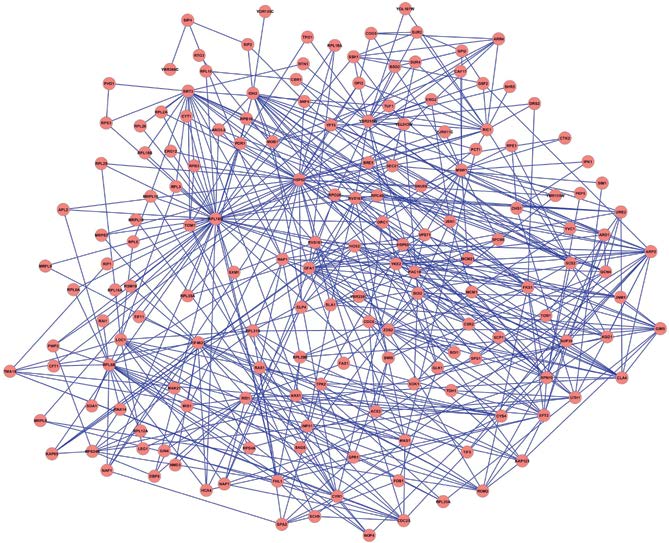
图1.26 酿酒酵母中调控寿命的基因/蛋白质网络。每个节点代表一个基因。(图10摘自Wimble C,Witten TM.2015.Interdiscip Top Gerontol 40:18–34.版权所有。2015.瑞士Basel的Karger出版社。经Karger许可。)
你在第五章中了解到,酿酒酵母至少有两个调节寿命的网络,这两个网络都与酵母的繁殖状态有关。繁殖寿命网络(replicative life span,RLS;图1.27A)调节具有繁殖活性的酵母的寿命。按时间顺序的寿命网络(chronological life span,CLS;见图1.27B)以寿命天数测量,调节非分裂或非生产性细胞的寿命。此外,最近的调查(见第5章)表明,RLS和CLS细胞的寿命可能会受到TOR网络的影响(见图1.27C)。事实上,当使用来自所有三个网络的数据生成一个图时,可以看到三个网络之间的显著交互(见图1.27D)。 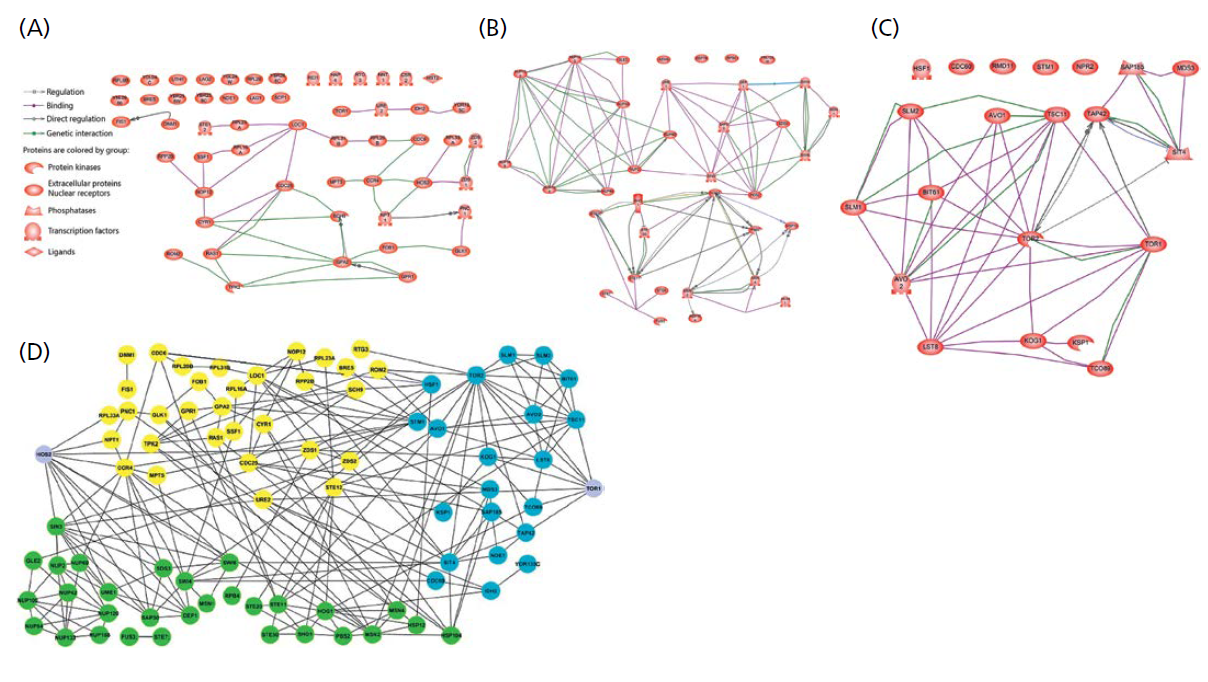
图1.27 与酿酒酵母寿命相关的部分网络。(A) 繁殖寿命;(B) 按时间顺序的寿命(CLS);(C)雷帕霉素(TOR)的靶点。(D) 包括RLS、CLS和TOR网络的长寿网络。A中的图例(legend)键也属于B和C。A、B和C中未连接的节点称为岛。岛是网络中尚未建立连接的潜在部分的节点。(D)中所示的蛋白质(节点)根据其独立网络进行颜色编码;黄色,RLS;绿色,CLS;蓝色,TOR。(图2、3、4和5摘自Wimble C,Witten TM.2015.Interdiscip Top Gerontol 40:18–34.版权所有,2015.瑞士Basel的Karger出版社。)
网络可视化是网络分析的重要一步。但是,需要统计度量计算来确定重要节点和边。尽管用于生成网络图的软件提供了多种方法来测量节点之间的关系,但在统计描述整个RLS网络中的连接时,我们只使用了更重要的测量方法之一,即中心性(centrality)(见图1.27)。中心性通过计算连接数或度数来衡量网络中节点的相对重要性。中心性还测量节点之间连接的强度,并回答“高度数节点是否与其他高度数节点有连接”的问题。节点的中心性越大,节点对网络的重要性就越大。
图1.28显示了连接到RLS网络的中心性数值。四个节点,每个节点对应一个特定基因,其中CYR1、GPA2、CDC25和SCH9的中心性数值最高。让我们更仔细地看看CYR1和GPA2基因,以便为中心性的使用提供意义。我们正在应用RLS网络的中心性分析其中的一种解释。我们在这里提供的单一解释只是为了便于介绍中心性的使用。当我们搜索酿酒酵母基因数据库时(https://www.yeastgenome.org/),我们发现CYR1基因编码腺苷酸环化酶,腺苷酸环化酶是将细胞外信号(例如出现营养)转移到细胞活动中的关键酶(见图1.25)。GPA2编码细胞和核膜受体的一部分,该受体结合营养物质,主要是葡萄糖。网络分析的结果有力地推断,营养代谢可能对酿酒酵母的寿命调节很重要。或者,RLS网络中相互作用的集成性质似乎是通过营养信号调节寿命。您在第5章中了解到,应用于RLS的定量分析支持实验观察,即减少喂给酿酒酵母的葡萄糖量可以延长寿命。 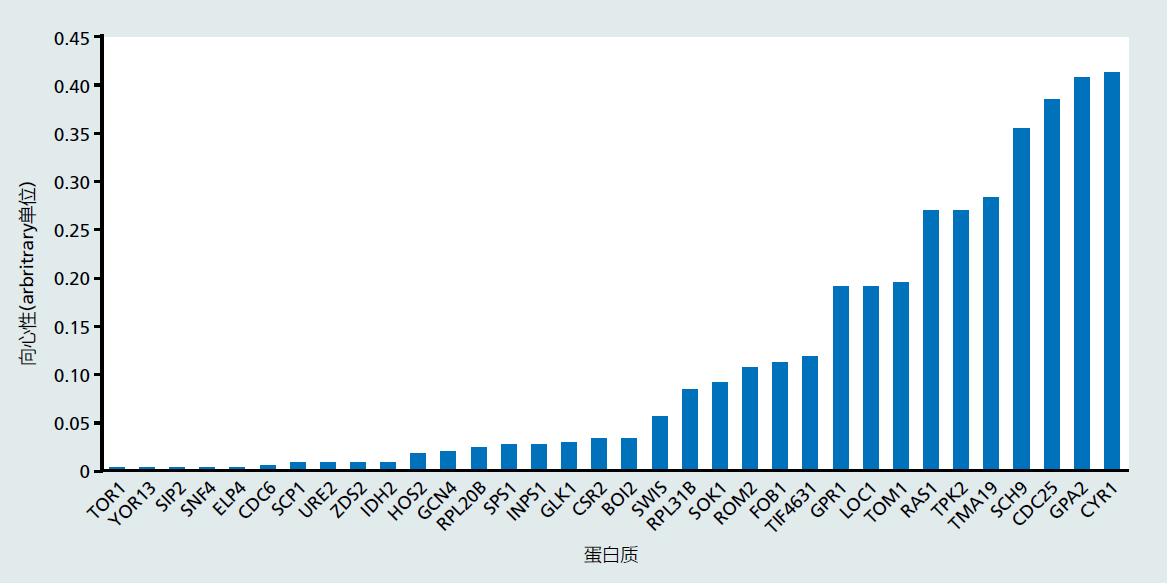
图1.28 图1.26所示蛋白质的中心性的计算。(摘自Wimble C,Witten TM.2015.Interdiscip Top Gerontol 40:18–34.经许可。版权所有。2015.瑞士卡格出版社。)
利用我们对中心性度量的解释,我们建立预测方程、算法或两者的下一步是收集更多关于CRY1和GPA2生物学行为的信息(见图1.23)。也就是说,回到实验室进行更多的实验,研究各种因素如何影响与这两个基因相关的生物化学和生理学过程。此外,还需要对人类的同源基因进行实验,以验证人类中存在类似的网络。这种网络分析和实验室实验的循环可能会持续几年到几十年,直到确定了调节寿命的绝对关键因素。假设到那时生物定律和常数也已经确定,那么就可以推导出一个预测寿命的方程式。