
第 2 部分疾病状态下的免疫系统
8 免疫病理学:免疫系统的故障
8.1 免疫病理学
到目前为止,我们一直在关注免疫系统的好处,即保护我们免于疾病的感染。
然而,免疫系统偶尔也会出现故障,有时甚至带来致命的后果。在这一讲里,我们将分析四类主要由免疫系统造成病理损伤的疾病。首先,讨论由免疫系统行使正常功能导致病理改变的疾病;接着,分析因免疫反应控制系统不能正常行使功能而引发的疾病;之后讨论因自身耐受机制破坏导致的自身免疫病;最后,集中讨论由遗传及后天免疫缺陷导致的疾病。
8.1.1 正常免疫反应引发的病理状态
结核就是一个由正常免疫系统功能导致病理性疾病的很好的例子。结核通常是因吸入感染者咳嗽时喷出的含有结核杆菌的微粒而感染的。当结核杆菌进入肺部后,它们将遭遇到守卫在那里制备截击呼吸道入侵者的巨噬细胞。
大家应该记得,在第一讲中提到的巨噬细胞首先通过一种叫吞噬体的囊泡来摄入入侵者。随后吞噬体被吞入巨噬细胞,并与另一种叫溶酶体的囊泡融合,溶酶体中含有多种能有效破坏细菌的化学物质。
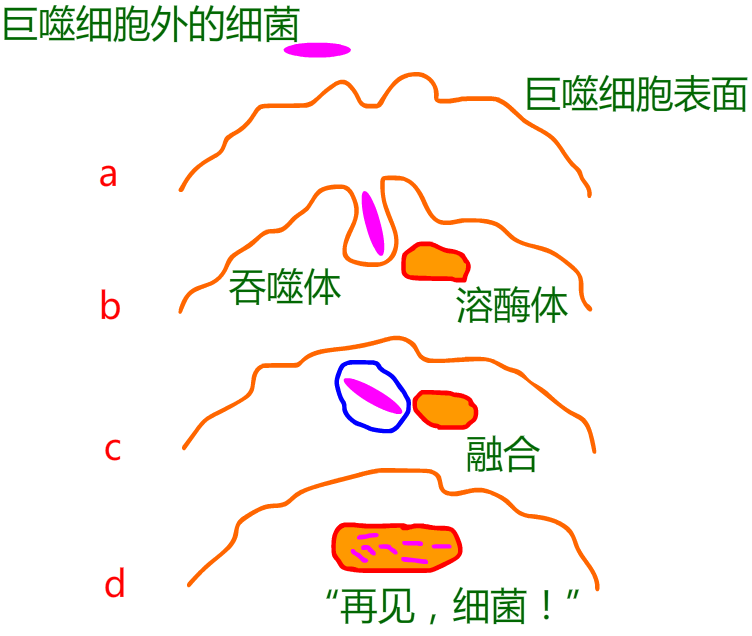
不幸的是,对于结核杆菌而言,巨噬细胞仅能将其吞入却不能将整个菌体消化掉,这是因为结核杆菌能对吞噬体表面进行修饰,使得吞噬体不能与溶酶体融合,从而结核杆菌在吞噬体内很安全,并且还能轻易得到其生长繁殖需要的营养,最终,大量新繁殖的结核杆菌撑破巨噬细胞释放出来,并使巨噬细胞死亡,而这些新生的结核杆菌继续感染附近的巨噬细胞。巨噬细胞坏死后,其溶酶体内容物释放到肺部组织,造成组织损伤并引发炎症反应,并将其他免疫细胞募集到该部位,导致更为严重的组织损伤。
巨噬细胞与结核杆菌之间的对抗导致“战斗性”细胞因子的产生,这些细胞因子继而使肺部巨噬细胞超激活。超激活的巨噬细胞杀伤能力大大增强,因而能更好地杀灭结核杆菌。然而,超激活的巨噬细胞释放的部分化学物质同样也能导致肺部组织损伤。
有时,激活的巨噬细胞以及由其所募集的细胞赢得这场战争并消灭入侵的结核杆菌,但有时候这场战争却是一个平局,形成慢性炎症,结核杆菌受到抑制,而巨噬细胞却持续被杀死,肺部也因炎症反应而持续受到伤害。因而在结核感染中,其病理表现恰是由巨噬细胞履行其职责——吞噬入侵者并召集其他免疫细胞参与战斗——导致的。
脓血症是另一种由免疫系统履行其职责导致的疾病。脓血症是个很一般性的叫法,描述了由系统性感染引发的症状,此类感染通常是由于作为我们身体第一道防线的物理屏障被突破后细菌进入血液而引起的。健康人患脓血症时,通常会有大量的细菌进入体内,例如细菌可以从脓肿或其他局部感染部位转移而进入人体引发感染。而对于免疫抑制患者,只需很少量细菌就可以引发感染了。
尽管革兰氏阴性菌和革兰氏阳性菌都能引起脓血症,但典型的元凶却是革兰氏阴性菌,例如大肠杆菌,其细胞壁组分中含有脂多糖(LPS),并能分泌 LPS 至周围环境中。正如我们在第二讲中所讨论,LPS 是一个强危险信号,能活化巨噬细胞和 NK 细胞,这两种细胞通过一个正反馈环路相互作用而增强彼此的激活状态,上调细胞因子的产生,并从血液中募集嗜中性粒细胞以及更多的巨噬细胞和 NK 细胞。
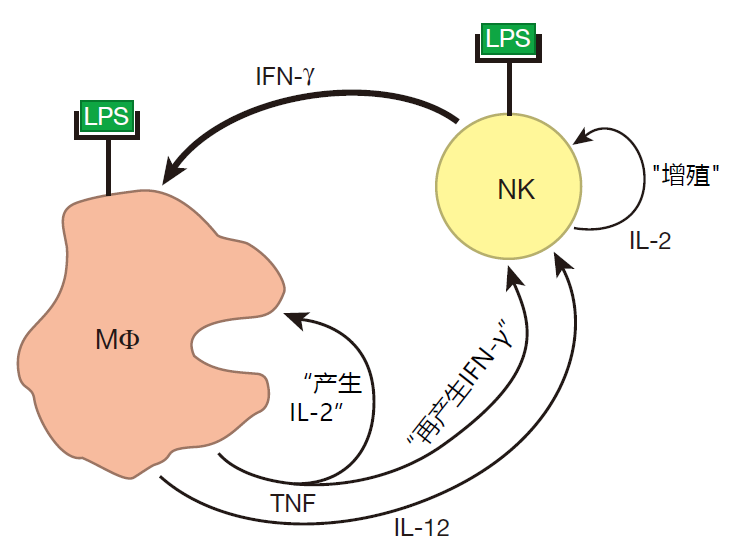
正常条件下,该正反馈环路的功能是增强免疫反应,使先天性免疫系统能够有效地对局部感染做出反应。然而在系统性感染中,细菌随血液进入人体各组织,增强了机体的反应但却帮了倒忙。激活巨噬细胞分泌的 TNF 使血管产生“漏洞”,导致血液从血管漏出进入周围的组织。在某些及其严重的情况下,因全身性渗漏引起的血量下降能导致血压降低,引发休克和心力衰竭。因而,虽然正常情况下这类正反馈循环能使先天性免疫系统强烈而迅速地对细菌入侵产生反应,但发生系统性感染时,正反馈循环却会引发机体的过度反应,从而导致脓血症和败血性休克的发生。
8.1.2 免疫调节缺陷导致的疾病
大约 1/4 的美国人对吸入或摄入的普通环境抗原(过敏原)过敏。非过敏体质者的免疫系统对过敏原的反应较轻微,主要是产生低水平的 IgG 抗体。与此形成强烈对比的是,过敏体质者(称为特应性个体)产生大量的 IgE,其血液中IgE 水平甚至能较非过敏体质者高出 1000-10000 倍。正是由于针对无害环境抗原产生过量的 IgE 导致了过敏。
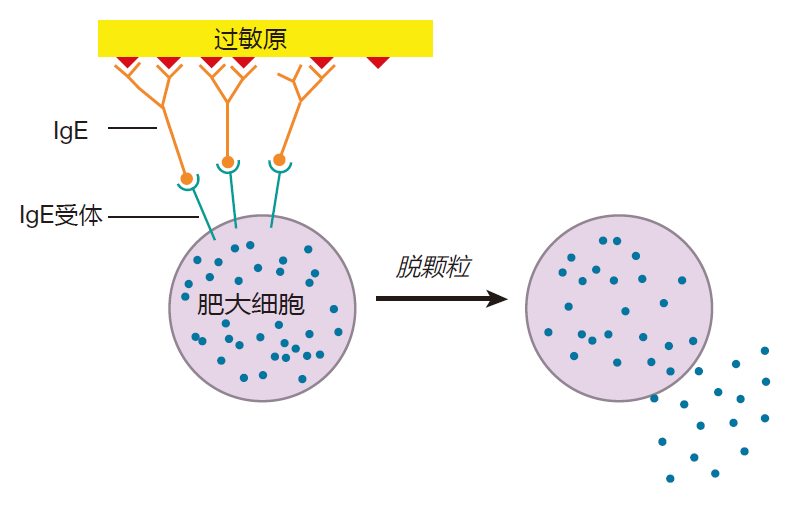
在第三讲中,我们讨论了 IgE 和肥大细胞的相互作用。由于肥大细胞脱颗粒是许多过敏反应的中心事件,现在让我们再来回顾一下这一过程。当特应性个体初次暴露于过敏原(例如花粉)时,体内产生大量能识别过敏原的 IgE。肥大细胞表面具有能结合 IgE Fc 区域的受体,因而在初次暴露后肥大细胞表面将结合大量过敏原特异性 IgE。过敏原是携带重复结构的小分子蛋白,IgE 能与其紧密结合。当第二次或更多次暴露于过敏原后,过敏原就能与肥大细胞发出“脱颗粒”的信号,使肥大细胞将正常情况下储存于内部的颗粒释放至其所在部位的组织。
肥大细胞的颗粒中含有组胺和其他非常有效的化学物质和酶,正是这些分子引发特应性个体极其相似的症状。
有趣的是,尽管 IgE 在血液中仅能存活约一天的时间,但一旦结合到肥大细胞上,IgE 的半衰期将长达数周。这意味着肥大细胞能随时保持“装备”,并在暴露于过敏原后较长时间持续脱颗粒。
过敏反应一般分为两种:速发相和迟发相。速发相反应由驻留于组织内的肥大细胞和嗜碱性粒细胞引起,后者可以由肥大细胞针对过敏原作出反应而释放出信号后从血液中募集到。和肥大细胞一样,嗜碱性粒细胞表面也有 IgE 受体,这些受体交联将导致嗜碱性粒细胞只能在组织中存活几天。
尽管肥大细胞和嗜碱性粒细胞均可介导针对过敏原的速发反应,但还有第三类含颗粒的白细胞——嗜酸性粒细胞,在慢性过敏反应中起主要作用(如哮喘)。
机体未受过敏原攻击时,组织或血液中仅有少量的嗜酸性粒细胞,一旦过敏反应发生,辅助 T 细胞就分泌细胞因子如 IL-5,从骨髓募集大量的嗜酸性粒细胞,这些嗜酸性粒细胞又为过敏反应贡献力量。由于嗜酸性粒细胞必须从骨髓中转移出来,相对于肥大细胞和嗜碱性粒细胞的立即反应,嗜酸性粒细胞的作用相对较晚。
当然机体创造出肥大细胞、嗜碱性粒细胞和嗜酸性粒细胞并非为了给过敏体质者带来烦恼。正常情况下,这些细胞“按指示”脱颗粒的能力为机体提供了抵抗寄生虫(如蠕虫)感染的能力,这些寄生虫通常很大,以至于不能被专职吞噬细胞吞噬。
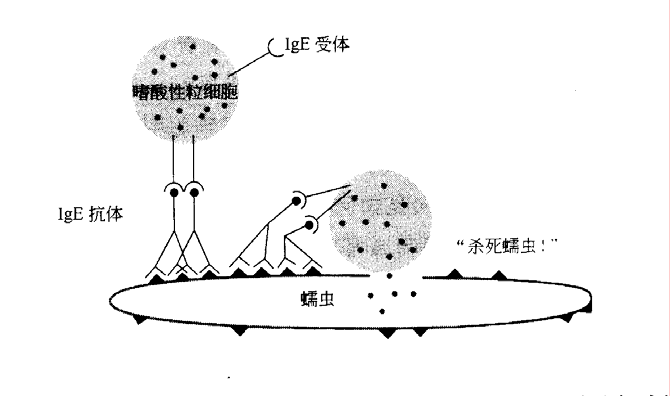
你可能已经注意到寄生虫(或过敏原)入侵肥大细胞/嗜碱性粒细胞的协同作用与细菌感染中巨噬细胞/嗜中性粒细胞的协同作用之间的相似之处。在这两种情况下,在组织中长期驻守的“哨兵”细胞对入侵者作出应答时被激活,进而从血液中召集一些短寿命的“雇佣兵”来协同作战。
目前已经清楚,对于过敏反应来说 IgE 是“坏分子”。然而究竟是什么决定了机体在过敏反应中应该产生 IgE,还是产生 IgG 呢?辅助性 T 细胞受到再刺激时会受环境因子影响分泌多种细胞因子,并且细胞因子种类会偏向 Th1 型或 Th2型。
这之后,根据生发中心内辅助 T 细胞产生的细胞因子种类,B 细胞将进行类别转换,产生 IgA、IgG 或 IgE,例如,若生发中心内以 Th1 型细胞为主,则 B细胞转换为产生 IgG 的类型,这是由于 Th1 细胞分泌 IFN-γ驱动 IgG 类转换。与此相反,若生发中心内以 Th2 型细胞为主并分泌 IL-4、IL-5,则 B 细胞转换为产生 IgE 的类型。因此,针对过敏原是产生 IgG 还是产生 IgE 主要决定于截击过敏原的二级淋巴器官内辅助 T 细胞的类型。与非过敏体质者相比较,过敏体质者体内辅助 T 细胞明显偏向 Th2 型。
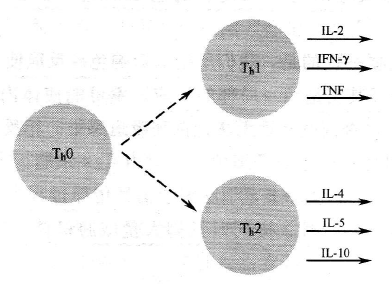
现在已清楚过敏体质者因其体内过敏原特应性辅助性 T 细胞偏向 Th2 型而
产生 IgE,那么这一倾向是如何形成的呢?对此目前尚无定论,但许多免疫学家认为 Th1/Th2 偏向通常在幼年时期形成,在某些个体,甚至于出生之前。下面就来讨论一下其形成过程。
胎儿的遗传物质大约一半来自母亲,一半来自父亲,因此胎儿实际上是一个移植体——其体内表达许多母体的免疫系统不耐受的父系抗原。由于胎盘是胎儿与母体之间的连接点,而胎盘内存在许多父系抗原,因而必须有措施来确保胎盘不被母体的 CTL 和 NK 细胞攻击。Th1 型辅助 T 细胞分泌 TNF 和 IL-2,TNF 能辅助活化 NK 细胞,IL-2 能刺激 NK 细胞和 CTL 增殖,因而,使母体的辅助 T细胞偏离 Th1 细胞因子谱分化对胎儿的存活是有利的。而事实也正如此,胎盘细胞产生相对大量的 IL-4 和 IL-10,诱导母亲的辅助 T 细胞分化成 Th2 细胞。但这些胎盘细胞因子同样也能强烈地影响胎儿的辅助 T 细胞,由此导致人类出生时体内的辅助 T 细胞显著地倾向于产生 Th2 型细胞因子。
显然,辅助 T 细胞的分化偏向并不会持续终生,最终大多数人体内 Th1/Th2细胞群会趋向平衡。幼年时期微生物感染(如病毒或细菌)可能有助于建立Th1/Th2 细胞群平衡,因为这些微生物感染通常引发 Th1 反应。但目前尚未确证早期微生物感染在对免疫反应进行“重排序”,使机体对过敏原产生 Th1 反应有重要作用。免疫学家猜测幼儿在感染了微生物并强烈地诱使其免疫应答偏向 Th1类的同时,如果接触到过敏原(如尘螨蛋白),针对该类过敏原的辅助 T 细胞应答同样也会偏向 Th1 类。一旦偏向发生,反馈机制将趋向于锁定 Th1 型反应,所产生的记忆 T 细胞不仅会记住过敏原,而且也会记住针对该类过敏原的 Th1类反应。一旦形成大量偏向记忆细胞,将很难纠正,因而在建立针对环境过敏原的正常免疫应答过程中,早期暴露于传染性疾病可能具有关键性的作用。
免疫偏向概念与发达国家过敏病例增加而微生物感染(如结核)降低的情况相符,因而有时也被称为“卫生学假说”。而儿童免疫偏向易感因素的存在也能解释在一年中特定月份出生的儿童更易于发生季节型过敏反应。
除环境因子(如早期暴露于传染病)外,遗传因素显然也在过敏易感的形成中具有重要的作用。例如,假如同卵双生子中一方有过敏症,则另一方患过敏的可能性为 50%。免疫学家已经指出针对某些过敏原过敏的人比不过敏的人更有可能遗传有特异的 MHCⅡ类基因,提示这些 MHC 分子可能在过敏原的提呈方面尤其有效。此外,部分过敏体质者体内产生 IgE 受体的突变体,据猜测这些突变体交联时发出非同寻常的强烈信号,导致肥大细胞分泌的 IL-4 水平的升高,反过来又刺激 IgE 的产生,在部分过敏体质者体内已经检测到 IL-4 基因启动子区有突变,并由此可能导致 IL-4 水平的升高。不幸的是,过敏易感基因难以确定,因为这些基因似乎数量较多,并且不同过敏体质者之间的易感基因还存在差异。
目前对此最佳的解释是过敏的免疫学基础在于免疫调节缺陷,致使过敏原特异性的辅助 T 细胞偏向表达 Th2 类细胞因子谱,并导致过敏原特异性 IgE 产生。
个体所遗传的基因可以使自身或多或少对过敏原易感,并且暴露于诸如微生物感染之类的环境因子可能影响易感个体是否成为特应性体质。
8.1.3 自身免疫病
机体并未建立一个完美的系统来仔细检测每一个T细胞和B细胞是否对自己产生耐受,相反,通过一套多层次的系统,该系统中每一个层次都有清除大多数自身反应性细胞的机制,而在前一层次中经过耐受诱导逃脱的细胞会被后一层次所捕获。一般情况下,这一机制运行良好,但偶尔也会出错导致系统被破坏。当维持自身耐受的机制受到严重破坏并足以产生病理状态时,自身免疫性疾病就发生了。很不幸,大约有 5%的美国人患有某种形式的自身免疫性疾病。
自身免疫紊乱可能是由遗传缺陷导致。例如大多数自身免疫病是包含自身反应淋巴细胞反复活化过程的慢性紊乱。在正常人体内,这一过程可通过活化诱导的细胞凋亡加以控制,当接受慢性刺激的 T 细胞表面的 Fas 蛋白与其配体结核后,T 细胞就可以被清除。Fas 或 Fas 配体有遗传缺陷的人缺乏这一层次的耐受保护,因而当其体内的 T 细胞受到自身抗原的持续刺激时并不会死亡。由此导致的疾病——自身免疫性淋巴组织增生综合症和 Canale-Smith 综合症,病理表现为大量的淋巴结肿胀,产生识别自身抗原的抗体,并在二级淋巴器官积聚大量的 T 细胞。
虽然部分自身免疫病是由遗传缺陷引起的,但大部分自身免疫病发生于耐受诱导机制无法清除自身反应性细胞的遗传正常的个体中,事实上,你可以认为自身免疫病的潜在可能性正是我们为拥有如此众多能识别如何入侵者的 T 细胞、B细胞受体而付出的代价。
目前最新的看法认为自身免疫病的发生至少需满足 3 个条件。第一,个体必须表达能有效提呈自身抗原肽的 MHC 分子。这意味着你所遗传的 MHC 分子在决定自身免疫病易感性时起主要作用。例如,仅有 0.2%的美国人患有青少年糖尿病,然而在遗传有两类 MHCⅡ类基因的高加索美国人中,患该病的可能性增加了大约 20 倍。
自身免疫病的第二个必要条件是患者体内需存在表达有能识别自身抗原受体的 T 细胞,在某些情况下还产生同样的 B 细胞。意味 TCR 和 BCR 是通过混合和匹配机制产生的,每一个体内的受体库跟别的个体都不相同,并伴随淋巴细胞的死亡或更新而改变,即便是同卵双生子所表达的 TCR 和 BCR 也不相同。
因此,在很大程度上,个体产生的淋巴细胞携带能识别特应性自身抗原的受体是很偶然的。
自身免疫病的发生的第三个必要条件是必须存在能打破清除自身反应性淋巴细胞正常耐受机制的环境因子。很多年以来,生理学家注意到,自身免疫病经常发生于细菌或病毒感染后;现在免疫学家认为相应的微生物感染可能是引发自身免疫病的关键性环境因子之一。目前已经很清楚病毒或细菌并不是引发自身免疫病的全部因素,因为对于大多数人来说,病毒或细菌感染并没有导致自身免疫病。但是在具备了遗传易感因素(如所携带的 MHC 分子型别)以及携带具有自身反应潜能受体的淋巴细胞之后,微生物感染就可能成为引发自身免疫病的导火线。
8.1.3.1分子模拟(molecular mimicry )
目前,在免疫学家中流行用于解释为什么感染可能破坏自身耐受机制的假说是“分子模拟(molecular mimicry)”,现在让我们来看一下具体内容。
淋巴细胞的 BCR 或 TCR 能识别同源抗原,但研究发现BCR或TCR并非仅识别单一抗原分子。诸如 MHC 分子能提呈大量具有相同特征(长度、结合基序等)的肽一样,一个 TCR 或一个 BCR 分子通常能识别数种不同的抗原。一般来说,一个 TCR 或一个 BCR 分子仅对这些同源抗原中的某一种或某几种具有高亲和力,而对其余抗原的亲和力相对较低。
微生物入侵时,受体能识别该微生物抗原的 B 细胞或 T 细胞被激活。分子模拟假说认为这些受体有时也会识别自身抗原,假如这样的识别发生,就会导致自身免疫病。研究人员猜测在感染微生物之前,那些具有自身反应潜能的淋巴细胞尚未被激活,因为它们的受体对自身抗原的亲和力过低而不足以启动激活,这也可能是由于原初淋巴细胞受转运模式的限制而不能接触到足够的自身抗原,从而不能被激活,然而,一旦与微生物抗原交叉反应后被激活,这些自身反应淋巴细胞就能职称真正的损害。例如。风湿性心脏病可能是喉部链球菌感染的并发症,其形成机制在于当识别链球菌抗原的辅助 T 细胞与提呈于心脏二尖瓣组织表面的蛋白发生交叉作用后,这些交叉反应性辅助 T细胞就可能引发炎症反应并对心脏瓣膜造成严重伤害。
人类自身免疫病的动物模型对于连接免疫系统哪些部分参与了自身免疫反应,哪些自身抗原是免疫反应的靶分子以及什么微生物抗原可能通过分子模拟机制引发自身免疫病是十分有用的。通常,这些模型中使用的动物应是经选育后对自身免疫病异常敏感,或是经基因改造后对自身免疫病易感。来自动物模型和人类自身的经验表明,识别自身抗原的 TCR 能和环境中的多种抗原发生交叉反应。
因此,尽管病毒或细菌感染可能成为某些自身免疫病的环境驱动因子,但任何一种自身免疫病均不太可能由一种微生物引发。
8.1.3.2 炎症和自身免疫病
尽管分子模拟可能活化起初漠视自身抗原的淋巴细胞,但仅此还不够。毕竟当自身反应性 T 细胞被进入组织的微生物分子拟态激活后,它们处于较危险的环境中。为了避免因忽视而引发的凋亡,自身反应性 T 细胞必须被持续再刺激,它们在环境中遇到的自身抗原不能提供足够的共刺激信号,就会导致免疫无应答或被清除。
大家还记得,通常由先天免疫系统给予获得性免疫系统行使功能的许可证,其机制部分在于先天性免疫系统细胞分泌的炎症细胞因子,如 IFN-γ和 TNF,激活 APC。一旦被激活,APC(如巨噬细胞)将表达 MHC 和共刺激分子对进入组织的 T 细胞进行再刺激。这意味着当淋巴细胞进入组织参与到先天性免疫系统对抗病原体的战斗时,其再刺激将不成问题。然而对于识别自身抗原的 T 细胞而言,即便先天性免疫系统并未把这些自身抗原视作危险,组织却可能是一个不友好的地方,因为这些自身反应性 T 细胞在这里通常得不到足够的共刺激信号来维持其生存。
这当中的关键在于,仅通过微生物的分子模拟尚不足以激活自身反应性 T细胞,同时还必须在表达自身抗原的组织中发生炎症反应。否则,自身反应性淋巴细胞并不会从血液进入组织,一旦它们进入组织,它们就可以存活。对于炎症反应的需求或许能解释为什么细菌感染(如喉部链球菌感染)很少导致自身免疫病(如风湿性心脏病)。
因而,大多数免疫学家所支持的自身免疫病的引发场景是这样的:首先是某个遗传易感个体感染了微生物,从而引发受体与自身抗原交联反应的 T 细胞被激活。与此同时,表达自身抗原的组织中发生了炎症反应。该炎症反应既可以由模拟的微生物自身引起,也可以由别的感染或创伤引起,之后,APC 被激活,并对自身反应性 T 细胞进行再刺激。此外,炎症反应所产生的细胞因子能上调表达于该组织中正常细胞表面的 MHCⅠ类分子,让这些细胞更易成为自身反应CTL 的靶细胞。
8.1.3.3 自身免疫病病例
自身免疫病通常分为两类:器官特异性和多系统性疾病。下面让我们来看看两类自身免疫病的例子,尤其要注意自身免疫反应所针对自身抗原以及可能参与了分子模拟的环境抗原。
胰岛素依赖性糖尿病就是一个很好的器官特异性自身免疫病例子。该疾病发生时,自身免疫病攻击的对象是胰腺的胰岛素β细胞。尽管由自身反应 B 细胞的抗体可能参与了导致糖尿病病理表现的慢性炎症,但目前认为对β细胞攻击的起始是由 CTL 介导的。
如果同卵双生子中一方患糖尿病,则另一方患病的可能性高达 50%,这一事实清楚表明存在着决定糖尿病易感性的遗传因素。到目前为止,尚无充分证据说明存在引发对β细胞产生攻击的环境因子。然而,许多免疫学家相信糖尿病的发生至少部分是由控制自身反应 CTL 的“调节性 T 细胞”没有发挥正常功能。不幸的是,CTL“受控制”在这里究竟意味着什么以及这些调节性 T 细胞到底是如何控制 CTL 的尚不清楚,还有待进一步研究。
糖尿病患者胰腺中胰岛素分泌细胞的破坏通常发生于糖尿病病症初次出现前数月甚至数年,因而糖尿病饮食也被称为“沉默的杀手”。幸运的是β细胞抗原的抗体在糖尿病早期就产生了,这样就可以通过检测糖尿病患者亲属体内是否产生这些自身反应性抗体来判断患者亲属是否处于糖尿病的早期。
重症肌无力是由自身抗体结合到一类重要的神经递质-乙酰胆碱受体上时引发的疾病。当正常情况下由乙酰胆碱所携带的信息不能由神经传递到组织时,肌肉就会变得无力并导致瘫痪。免疫学家已经注意到脊髓灰质炎病毒蛋白一个区域的氨基酸序列与乙酰胆碱受体的部分区域相似,表明脊髓灰质炎病毒感染可能提供了受体可与乙酰胆碱受体受体交叉反应的淋巴细胞活化时所需的模拟分子。
多发性硬化症是一类由自身反应性 T 细胞引发的中枢神经系统的炎症性疾病。在多发性硬化症中,慢性炎症反应破坏了脑部神经细胞有效传递电信号所需的髓鞘,导致感觉输入(如视觉)缺陷及麻痹。由 T 细胞分泌的细胞因子所募集的巨噬细胞在炎症反应中发挥主要作用。一开始并不清楚 T 细胞怎么会进入大脑并引发多发性硬化症,后来发现激活的 T 细胞能穿过血脑屏障,推测这些 T细胞的靶分子是髓鞘的一个主要成分——髓磷脂碱性蛋白。从多发性硬化症病人体内分离得到的 T 细胞即能识别髓磷脂碱性蛋白的衍生肽,也能识别由单纯疱疹病毒和 EB 病毒(该蛋白可引发单核细胞增多症)编码的蛋白衍生肽。因而可能的机制是当遗传易感个体感染了疱疹病毒或 EB 病毒后,体内产生识别病毒蛋白的 T 细胞,这些激活的 T 细胞一部分可能仅有能与髓磷脂碱性蛋白交叉反应的受体,一旦这些 T 细胞穿过血脑屏障,就能攻击髓鞘,从而引发多发性硬化症。
然而,因感染疱疹病毒和 EB 病毒而引发多发性硬化症的人很少,因而仅微生物分子模拟尚不能完整解释自身免疫病的发生。实际上,正如大多数自身免疫病,多发性硬化症具有强烈的遗传倾向——同卵双生子同患多发性硬化症的可能性是异卵双生子的 10 倍,而异卵双生子同患多发性硬化症的可能性是普通人的20 倍。此外,很有一些多发性硬化症发病率相对较低的“抗性”群体(如西班牙人、亚洲人、美洲土著),推测原因可能在于这些人群具有独特的遗传族谱。
寻常性天疱疮的产生机制是针对皮肤表面一个自身蛋白(桥粒核心糖蛋白Ⅰ)的抗体破坏了皮肤细胞间的黏附,从而导致皮肤产生水疱。事实上,将从天疱疮患者体内分离到的抗体注射到动物体内,能引发天疱疮症状。有趣的是,到目前为止,一个编码一种特殊类型 MHCⅡ类分子的基因仅在天疱疮患者体内发现,提示个体所遗传的 MHC 分子类型在决定天疱疮易感性中起重要作用。
类风湿性关节炎是以关节慢性炎症为特征的全身性系统性自身免疫病。据推测,该自身免疫反应的攻击目标之一是一种特定的软骨蛋白,并且自类风湿性关节炎患者体内分离的 T 细胞能识别软骨蛋白和一种由引起结核的细菌所编码的蛋白。就一点来说,注射分枝杆菌的小鼠出现关节炎症提示了,但不能充分证明部分患者的类风湿性关节炎可能是由结核杆菌引起的。
在类风湿性关节炎患者关节部位发现能结合 IgG 尾部的 IgM 抗体。这些抗体相互间能形成 IgM-IgG 抗体复合物,激活进入关节的巨噬细胞,加重炎症反应。
实际上,类风湿性关节炎相关炎症反应主要是由自身反应性辅助 T 细胞指引下浸润关节部位的巨噬细胞所产生的 TNF 引起。目前在关节炎的治疗中,主要使用两种能拮抗 TNF 的药物,一种是 TNF 抗体,能阻止 TNF 发挥作用,另一种是 TNF 的假受体,他们都能有效缓解类风湿性关节炎患者的症状。
红斑狼疮也是一种系统性自身免疫病,在美国约有 250000 名患者,其中90%是女性。红斑狼疮有多种表现形式,包括额头及脸颊部出现红色皮疹(该病名称由此而来)、肺部炎症、关节炎、肾脏损伤、脱发、瘫痪以及痉挛。狼疮的发生是由于 T 细胞、B 细胞耐受被打破,导致体内产生大量能识别包括 DNA、DNA-蛋白复合物、RNA-蛋白复合物在内的多种自身抗原的 IgG 抗体。狼疮患者所表现出的症状反映了体内自身抗体的种类。这些自身抗体形成自身抗原-抗体复合物,阻塞体内含有“滤器”的器官(如肾、关节、大脑),从而引发慢性炎症。
异卵双生子同患狼疮的概率大约为 2%而对于同卵双生子来说,这一概率增加了大约 10 倍。这表明该病明显具有遗传因素,目前已确定多个相关的 MHC以及非 MHC 基因,其中的每一个看起来都能略微增加患狼疮的概率。目前还没有发现特异性的微生物感染与狼疮之间的联系,但缺失功能性 Fas 基因或 FasL基因的小鼠表现出狼疮样症状。免疫学家据此推测狼疮的发生可能包含了激活诱导性细胞死亡的缺陷,即本该应慢性刺激而死亡的淋巴细胞却存活下来,由此导致了狼疮的扩散。
8.1.3.4 抗原的扩散
导致免疫学家难以确定引发自身免疫病的环境因子的原因之一在于抗原扩散现象。从自身免疫病患者体内分离的 T 细胞或 B 细胞通常能识别数种,有时甚至是许多种自身抗原。由此看起来尽管自身免疫反应最初可能只包含识别单一“起始”自身抗原的 T 细胞或 B 细胞,但随着时间的推移,识别其他自身抗原的淋巴细胞也会被激活。
举例来说,狼疮最初是由识别蛋白包被的 DNA(正如通常情况下人体内的DNA)的 T 细胞和 B 细胞引发的,这些蛋白包被的 DNA 通过感染或创伤从细胞中释放出来,一旦其释放出来后,自身免疫反应将导致大量的巨噬细胞和其他免疫细胞的聚集,并表达 IFN-γ和 TNF 等细胞因子,提高巨噬细胞和树突状细胞的抗原提呈效率,从而加重炎症反应。由此导致自受损组织释放的和之前因无充足的 MHC 分子或共刺激信号而提呈得很少得其他自身抗原能够有效提呈,并激活曾忽视其存在得活化 T 细胞,因而自身反应性 T 细胞得目标由最初得自身抗原扩散至原来因沉默而被忽视得其他自身抗原。
8.1.4 免疫缺陷病
当我们的免疫系统不能再全力以赴地工作时,就可能引起严重的疾病。部分免疫缺陷是由遗传缺陷所导致的免疫网络的某些部分障碍引起的,另外一些是由于营养不良、特异的免疫抑制(如器官抑制或癌症的化疗)或疾病(如 AIDS)
而导致的。
8.1.4.1 遗传缺陷导致的免疫缺陷
单基因突变引起的遗传缺陷能导致免疫系统衰弱。例如,生来就携带无功能CD40 或 CD40L 蛋白的个体不能产生 T 细胞依赖抗体反应,因为其 T 细胞不能传递至关重要的共刺激信号,而 B 细胞脱氧也不能接受这些信号。CD40-CD40L缺陷所导致的结果是 B 细胞分泌亲和力不成熟的 IgM 抗体,因为类别转换和 T细胞高突变均需要 CD40L 所传递的共刺激信号。
其他一些遗传缺陷能影响胸腺的形成。DiGeorge 综合症是该类缺陷的一种,在这种疾病中,几乎所有的胸腺组织都缺失,患此病的人因缺乏功能性 T 细胞而对某些致命感染易感。
遗传缺陷也能导致 T 细胞和 B 细胞的缺失,这类疾病被称为严重联合免疫缺陷病(SCID),其中联合表明 T 细胞和 B 细胞均功能失常。著名的“泡泡男孩”就是因为患该病而必须隔离在无病原环境中。尽管许多突变均能导致 SCIDS,但目前研究最为透彻的是一个能产生成熟 T 细胞、B 细胞受体所需基因剪接蛋白的缺陷。没有了成熟的受体,T 细胞和 B 细胞就“看不见”周围的环境,变得毫无用处。
免疫缺陷也可以由先天性免疫系统的遗传缺陷导致。例如,先天补体蛋白(如C3)缺陷的人伴有淋巴结构异常(无生发中心),且 B 细胞产生大量的 IgM 抗体。
尽管许多蛋白都参与维持系统性和获得性免疫系统的正常运作,然而令人惊异的是导致免疫缺陷的突变却很罕见。事实上,大约在 1 万个新生儿中仅有一个具有免疫缺陷。这可能是由于我们“丰裕”的免疫系统进化出了一套机制,在免疫系统主要组分失效时为其提供相应的备份,从而使得许多遗传性免疫缺陷无明显表现。
8.1.4.2 AIDS
遗传性免疫缺陷相对少见,成千上万的免疫缺陷都是获得性的。许多人是因感染了 HIV 而患免疫缺陷。目前全球已有超过 4000 万的人感染了 HIV。最初提示医生 AIDS 的是在他们医治免疫缺陷病人的过程中,发现这类病人通常表现为感染(如卡氏肺囊虫性肺炎)或癌症(卡波西肉瘤)的高发率,而这些疾病通常只在免疫抑制者中出现。之后,导致免疫缺陷的病毒被分离出来,并命名为 1型人类免疫缺陷病毒(HIV-1)。
HIV-1 感染的发生和其他病毒感染非常相似。起始感染的病毒进入人体细胞后利用细胞的生物合成机器大量复制自身的拷贝,新产生的病毒从细胞释放出来并感染其他细胞。因此在感染的早期阶段,由于先天性免疫系统已发挥其作用,而获得性免疫系统尚处于激活过程中,相对来说病毒的繁殖不易察觉。大约一周后,获得性免疫介入,病毒特异性 B 细胞、辅助 T 细胞和 CTL 开始激活、增生并开始发挥作用。此阶段为病毒感染的“急性”早期,由于病毒在受感染的细胞内繁殖而使体内病毒数量(病毒载量)显著升高。之后,病毒特异性 CTL 和抗体开始发挥作用,病毒载量明显下降。
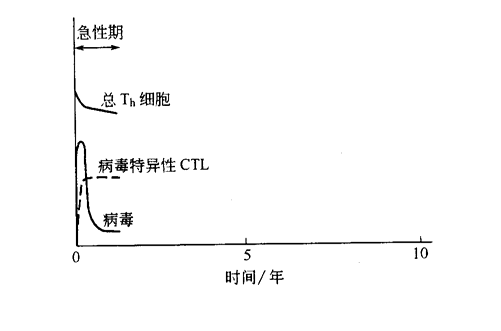
对于许多病毒(如天花)而言,急性期病毒感染的最终结果是“无菌”——所有入侵的病毒都被破坏,体内产生大量记忆 B 细胞和 T 细胞,以对抗以后同种病毒感染。然而仅有很少数的很幸运的人在感染 HIV-1 后也会以“无菌”而告终。
对于大多数人来说,HIV-1 感染能导致持续 10 年以上的“慢性”感染期,在此阶段,免疫系统与 AIDS 病毒之间展开激烈的对抗——很不幸的是,病毒通常会赢得这场战斗。
与急性期所达到的病毒载量相比,慢性感染期的病毒载量下降,而病毒特异性 CTL 和辅助 T 细胞仍维持在高水平,这表明免疫系统仍在努力抗击病毒。
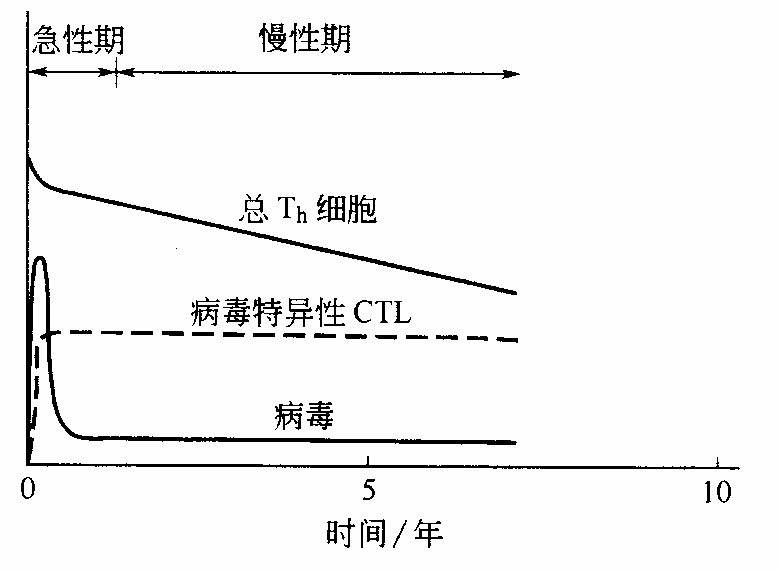
然而随着慢性期的进展,Th 细胞由于感染了病毒而被杀死,因此其总量缓慢下降,最终将无足够的辅助 T 细胞来提供病毒特异性 CTL 所需的帮助。这之后,CTL 数量也开始减少,而病毒载量则上升,这是因为没有足够的 CTL 来对付新产生的感染细胞。
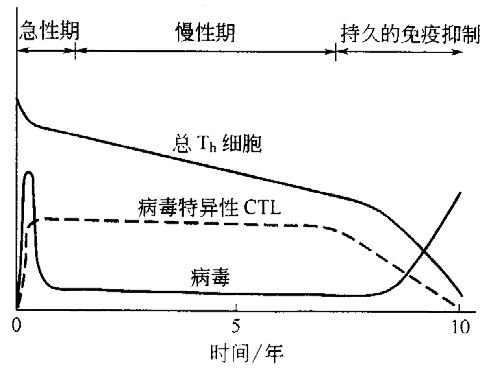
最后,免疫系统的防御彻底崩溃,所导致的免疫抑制状态使患者对病原体感染完全不设防,即便是那些对完好免疫系统个体不会造成如何伤害的病原体。更为糟糕的是,这些通常是“条件性”的感染对于免疫系统已被破坏的 AIDS 病人而言都可能是致病的。
为什么HIV-1能击败哪些曾成功保护我们不受其他大多数病原体侵害的免疫系统?答案有两个部分。首先是 ADIS 病毒本身。所有病毒基本上都是由遗传物质(DNA 或 RNA)及一个保护性的外壳组成。AIDS 病毒的遗传物质是 RNA,在进入靶细胞后,病毒 RNA 由其自身编码的逆转录酶逆转录为互补的 DNA(cDNA),接着,细胞 DNA 被病毒所携带的另一个酶切断,而病毒 cDNA 插入细胞 DNA 的间隙。这是最糟糕的,一旦病毒 DNA 插入细胞 DNA 中,病毒 DNA就会留在上面,病毒进入潜伏状态,之后受感染细胞就不能被 CTL 检测到。这之后一段时间,由于一些目前尚不清楚的信号反应,潜伏的病毒就会被再激活,产生更多的病毒拷贝,新生的病毒又再感染别的细胞。
因此,HIV-1 感染成为如此一个难题的一个重要特征就是 HIV-1 建立起潜伏状态使之不能被 CTL 检测到。但含有更糟糕的。用于复制病毒 RNA 的逆转录酶具有严重的出错倾向:每转录一条病毒 RNA 或 cDNA 就有一个错误(突变)产生。这意味着受感染细胞内产生的新病毒通常会和最初感染的病毒不同。突变的结果有三种,第一种突变不改变病毒的结构和功能;第二种突变能杀死病毒,因为突变阻断了病毒的某些基本功能(如突变可能造成逆转录酶改变,使之不能再复制病毒 RNA);第三种也是最可怕的一种,突变帮助病毒适应环境,使病毒更具有破坏性。
例如,突变可能造成一个病毒多肽的改变,使 CTL 不能对其进行识别,或是使得该多肽不能被 CTL 识别的 MHC 分子提呈。这样的突变使原来的 CTL 对感染了突变病毒的细胞变得毫无用处,而必须活化能识别其他病毒多肽的新的CTL。与此同时,逃过 CTL 监测的突变病毒疯狂复制,并且每感染一次新的细胞,病毒就发生一次突变。AIDS 病毒的突变率如此之高,使得病毒总是能领先抗击它的 CTL 或抗体一步。
因此,HIV-1 尤为致命的两个特征就是病毒能建立起无法被检测到的潜伏状态以及病毒的高突变。但这还只是故事的一半。HIV-1 的感染还能针对其他的特异靶细胞——辅助 T 细胞、巨噬细胞和树突状细胞。HIV-1 感染时,其所结合的受体蛋白是 CD4——大量存在于辅助 T 细胞表面的共受体蛋白。CD4 也可由巨噬细胞和树突状细胞表达,但这两类细胞表面的 CD4 分子较少。HIV-1 病毒攻击这些细胞,破坏细胞功能,杀死细胞,或使这些细胞成为 CTL 的攻击目标,因而激活 CTL 并为其提供帮助的是被 HIV-1 损伤或破坏的免疫细胞。
更为阴险的是,HIV-1 能通过运用免疫系统的基本功能来使免疫系统对付自身,从而扩散并持续病毒感染。例如,HIV-1 能结合到树突状细胞的表面并由这些细胞从 CD4+细胞相对较少的组织中转运至含有大量 CD4+细胞的淋巴结内。
淋巴结内不仅有大量的 CD4+T 细胞,并且大部分 CD4+细胞还处于增殖的过程中,这使它们成为 HIV-1 增殖的理想场所,对于 HIV-1 而言,淋巴结就是其天堂。
经抗体或补体调理后,HIV-1 仍能通过滤泡树突状细胞而保留在淋巴结中,这种提呈可以帮助激活 B 细胞,但 CD4+T 细胞也会经过这片 FDC 的丛林,从而被结合在树突状细胞上的 HIV-1 颗粒感染。由于病毒颗粒通常能结合在 FDC上数月之久,淋巴结实际上已经成为了 HIV-1 的仓库。因而通过选择性地感染CD4+T 细胞,HIV-1 能利用这些免疫细胞经淋巴结的正常转运途径,并把这个约会酒吧变成 HIV-1 自己的游乐场。
总之,HIV-1 感染的病理结果是由于病毒缓慢破坏患者免疫系统,引起严重的免疫抑制,从而最终导致机体死亡。病毒之所以能这么做是因为它能建立起一种潜伏的“秘密”感染机制,并具有高突变率以及优先感染并破坏正常情况下本该对抗病毒感染的免疫细胞。