
9.2 与年龄有关的人类大脑疾病:阿兹海默症和帕金森症
Compared with what scientists know about other human organs, they have only a rudimentary understanding of how the human brain performs its functions. We know even less about how aging affects brain function. Nonetheless, we do know that the healthy human brain seems to retain significant function and to show little structural change throughout most of the adult life span. In cases where changes have been reported, there is significant variation among individuals with respect to the amount of age-related dysfunction. Moreover, the anatomical location in the brain where age-related functional loss occurs also differs significantly among individuals. Thus, generalities about age-related functional loss in the human brain are difficult to make. You will see, however, that age-related alterations in the human brain, although minor, have the potential to progress to devastating neuropathology.
In this section, we focus exclusively on the two most common age-related diseases that affect the CNS: Alzheimer‘s disease and Parkinson‘s disease. (Age-related diseases of the PNS, the nerves that carry information to and from the CNS, have not been well studied and are not covered here.)
9.2.1 Changes in structure and neurotransmission seem to be minor in the aging brain
Structural changes in the brain at advanced ages are consistently found to be minor, to vary among individuals, and to not affect the same anatomical location in all people. Moreover, the ability of the brain to develop new neural connections (that is, its neuroplasticity) remains remarkably robust in the aging human brain. In fact, studies using magnetic resonance imaging (MRI) find only small changes in brain size with aging. The lack of brain atrophy reflects two general observations: (1) neural cell number remains fairly constant throughout the adult life span, and (2) glial cell formation slightly increases (in a process called gliosis) with age. When neural cell loss has been reported, it appears to be limited to specific brain centers such as the hippocampus, locus coeruleus, and cerebellum. The importance of age-related neural cell loss to brain function remains obscure, because slight neural cell loss does not correlate well with decreased function.
There also seem to be only minor age-related changes in the structures involved in neurotransmission, including synaptic density, synaptic size, and volume of the synaptic cleft. Although determining these values for large populations of aged individuals is technically difficult, and reports of a post-reproductive decline in synaptic quality have generally been limited to brains derived from autopsy, synaptic quality appears to be highly maintained, except in the hippocampus, where synaptic size and volume decline. However, functional loss due to changes in synaptic quality have not been demonstrated.
Current evidence also suggests that concentrations of neurotransmitters, such as acetylcholine, norepinephrine, epinephrine, and dopamine, do not decrease significantly with age. However, measurements can be done only indirectly in large populations by measuring neurotransmitter concentrations in blood and urine. New techniques using labeled probes in combination with imaging technology are being introduced, but the population of aged individuals in which these techniques have been applied remains too small for definitive conclusions.
9.2.2 Amyloid plaques and neurofibrillary tangles accumulate in the aged brain
The aging human brain appears to have a diminished capacity to adequately rid itself of damaged 蛋白质, and this results in an accumulation of potentially toxic compounds. Two of these neurotoxic compounds, amyloid plaques and neurofibrillary tangles, have been identified as precursors to neurological diseases such as Alzheimer‘s disease and Parkinson‘s disease (Figure 9.7) . However, accumulation of these compounds in the healthy aging brain does not seem to have a significant effect on functional capacity in the majority of post-reproductive individuals. Only about 10–20% of aged individuals will experience a transition from normal aging to definable neurological disease as a result of the accumulation of amyloid plaques and neurofibrillary tangles.
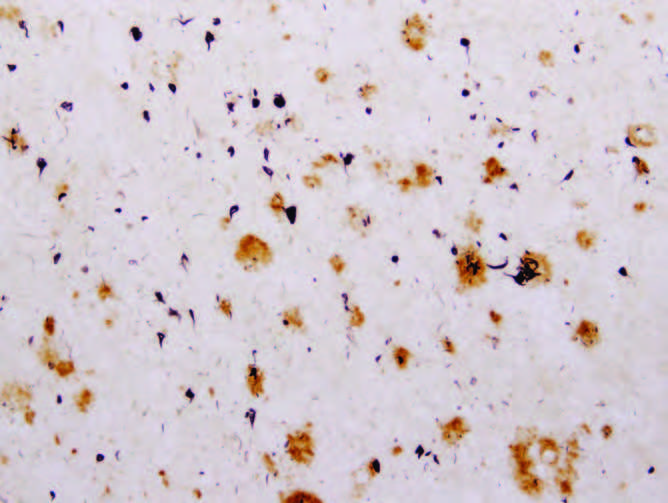
Figure 9.7 Amyloid plaques ( large circles ) and neurofibrillary tangles ( small circles ) in the brain tissues of an individual who had Alzheimer's disease. (From E.G. McGeer and P.L. McGeer, Mol. Interv. 1:22–29, 2001. With permission from American Society for Pharmacology and Experimental Therapeutics.)
More than 150 years ago, the father of modern pathology, Rudolf Virchow, described a “waxy substance” in the brain of older individuals that had the appearance of a starchlike compound. He named this substance amyloid. (The structure of the amyloid 蛋白质 is not related to the carbohydrate family, but the name remains.) For the next 130 years, detailed descriptions of amyloid plaques were published. Then, in 1984, the amino acid sequence of the 蛋白质 forming the amyloid plaques, Aβ 蛋白质, was elucidated. (The β designation refers to the 蛋白质's secondary structure, a β-sheet, a rigid structure formed by hydrogen bonding, as shown in Figure 9.8.) The amyloid fibrils (plaque) are made up of several Aβ 蛋白质 wrapped around each other to create a highly insoluble molecule. The formation of amyloid plaques from Aβ 蛋白质 in brain tissue is a normal part of human aging.
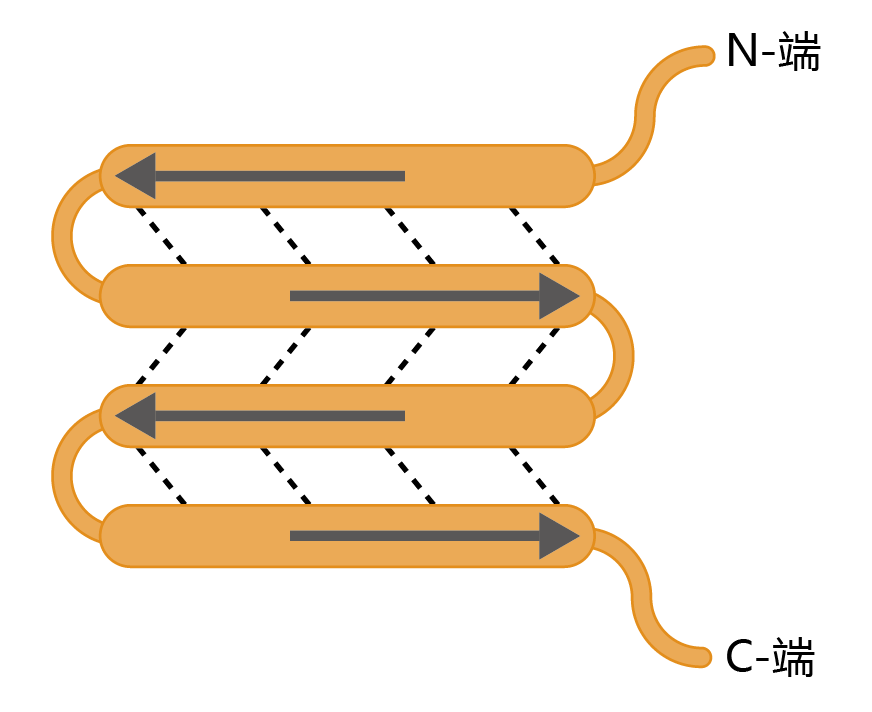
Figure 9.8 β-sheet secondary structure of a 蛋白质. Arrows indicate the direction (N-terminus to C-terminus) of the amino acid sequence. Dotted lines indicate the hydrogen bonds that hold the sheets together. Notice the antiparallel pattern of the hydrogen-bonded sequences. This configuration gives the β -sheet structure its rigidity.
Analysis of the amino acid sequence of the Aβ 蛋白质 led to the discovery of the gene that encodes the 蛋白质. The gene was found to be part of a larger gene, localized to Chromosome 21, that encodes the amyloid precursor 蛋白质 (APP). APP has a large hydrophilic extracellular domain, a transmembrane domain consisting of 23 amino acids, and a small intracellular domain (Figure 9.9) . The expression of APP occurs in both neural and nonneural tissue. Although the function of APP has yet to be fully elucidated, physiological studies suggest that APP supports dendrite outgrowth, synaptogenesis, and inhibition of platelet activation, and it may function as a copper transport 蛋白质. Synthesis of APP in neural tissue takes place in the endoplasmic reticulum, with post-translational modification occurring during transit to its position in the cell membrane. The processing of APP within the membrane can follow one of two proteolytic pathways (Figure 9.9). The non-amyloidogenic, or α, pathway produces the 蛋白质, p3 (3 kD), whose function and metabolism remain largely unresolved. The amyloidogenic, or β, pathway produces the Aβ 蛋白质. Enzymes known as secretases determine which proteolytic pathway a membranebound APP will follow. In nonneural tissue, α-secretase predominates, resulting in the production of the nontoxic p3 蛋白质. In contrast, in neural tissue, β-secretase (also known as β-site APP cleavage enzyme 1, or BACE1) predominates, resulting in formation of the potentially neurotoxic Aβ 蛋白质.
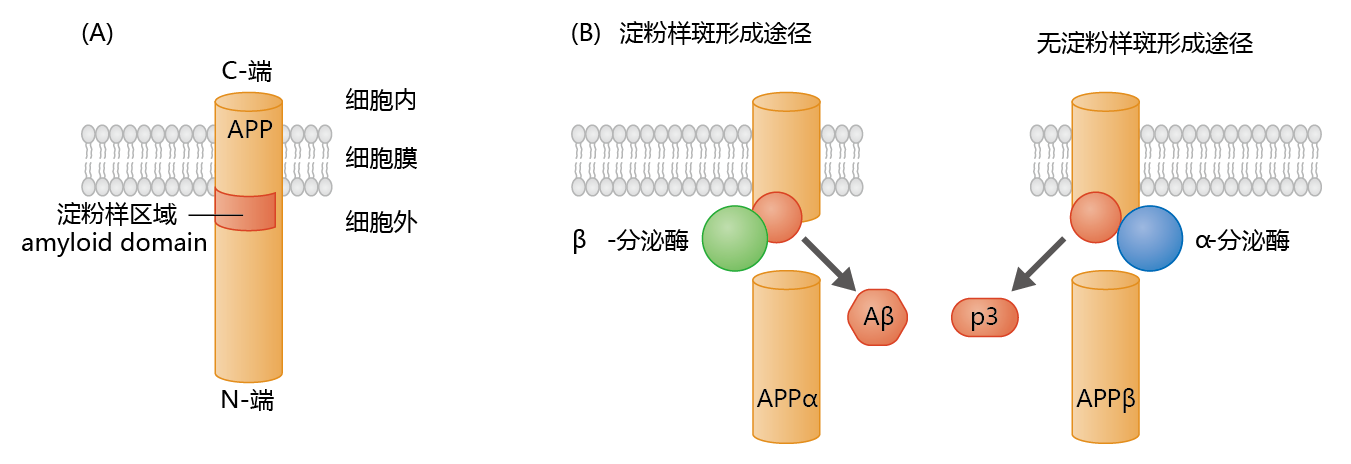
Figure 9.9 The amyloid precursor 蛋白质 (APP) and formation of the Aβ 蛋白质. (A) APP contains an amyloid domain (40–42 amino acid residues) that is mainly located in the extracellular space, with just a small hydrophobic region within the membrane. (B) β -secretase, an enzyme found primarily in neural tissue, cleaves APP just below the amyloid domain. This leaves a small intramembrane 蛋白质 with a conformation favoring formation of the A β 蛋白质. Cleavage of APP by α -secretase, the enzyme found primarily in nonneural tissue, favors the formation of the nontoxic p3 蛋白质.
Production of Aβ in neural tissue leads to the formation of amyloid plaque, a highly insoluble structure with no known catabolic pathway (Figure 9.10) . The pathway from Aβ to amyloid plaque has not been described in detail, although a general theory has begun to emerge. It appears that Aβ polymerizes through sequential formation of dimers, tetramers, and long oligomers. The oligomers aggregate into a stacked conformation, known as a β-cross formation, forming a fibril. Several fibrils then aggregate to form the plaque. Enzymes catalyzing Aβ polymerization have not been identified, suggesting that aggregation is nonenzymatic and concentration dependent. Aβ amyloid plaques are found primarily in the extracellular space, as would be predicted from the APP-processing mechanism. However, recent studies have found low concentrations of intracellular Aβ amyloid plaques in the Golgi complex and endoplasmic reticulum.
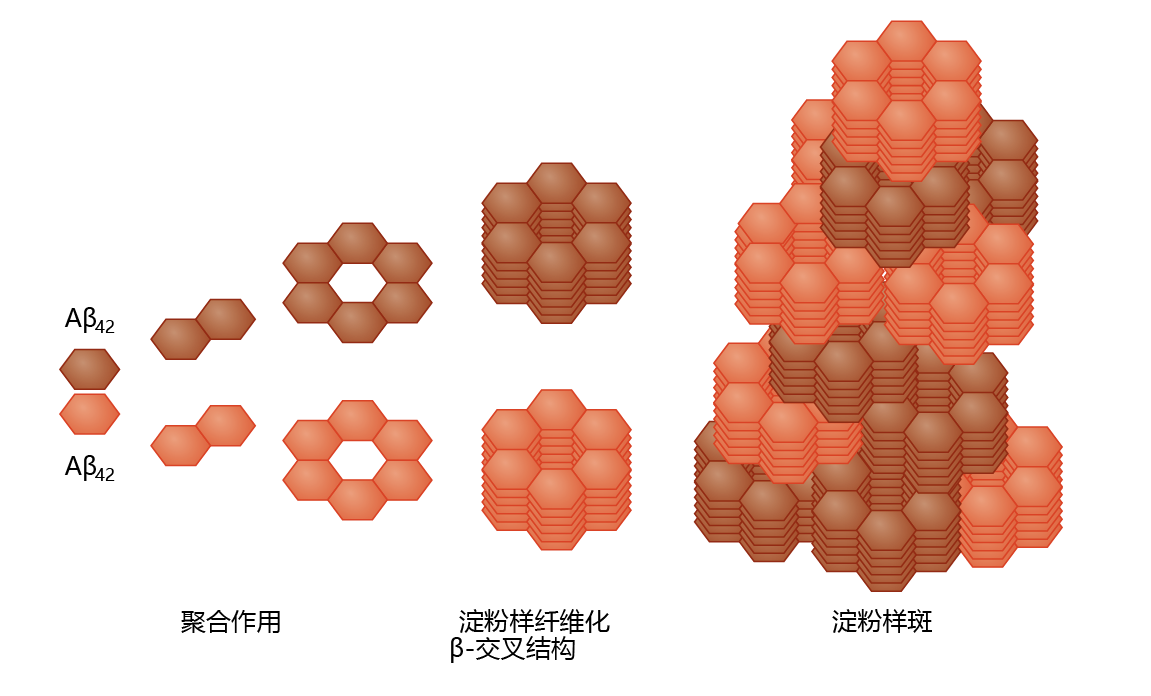
Figure 9.10 Amyloid plaque formation. Left to right: At high concentrations, the A β 42 蛋白质 cleaved from the APP complex (see Figure 9.9) forms aggregates by sequential polymerization. The polymerization process leads to the formation of a fibril in the β -cross configuration—a configuration that resists proteolysis. This induces further aggregation into the stacked configuration of the amyloid plaque.
The limited understanding of the mechanism underlying amyloid plaque formation reflects the unique ability of Aβ to form aggregates. The formation of 蛋白质 aggregates in biological systems is the exception rather than the rule and most likely reflects an abnormally folded precursor 蛋白质—in the current scenario, the Aβ 蛋白质. The cell invests considerable energy into ensuring that the folding process occurs without error and independent of other 蛋白质, a critical control mechanism needed to maintain the structure-function relationship of 蛋白质 activity. Several intracellular 蛋白质, many of which are part of the chaperone family, support the 蛋白质-folding process by inhibiting aggregate formation or by molecularly marking for degradation those 蛋白质 that have formed an aggregate. Such chaperone 蛋白质 do not seem to exist for the Aβ 蛋白质, resulting in an aggregate—that is, the amyloid plaque—that resists proteolysis.
Neurofibrillary tangles are insoluble twisted fibers found inside brain cells. The microtubules of the cytoskeleton perform a vital function by directing the movement and determining the final position of organelles and/or 蛋白质, from the cell body to the axon. Under normal conditions, binding of the tau 蛋白质 to the microtubule provides stability to this cellular structure (Figure 9.11) . The 蛋白质's function as a support 蛋白质 depends on its degree of phosphorylation, a process regulated by various kinases and phosphatases. Normal tau has 2–3 moles of phosphate per mole of 蛋白质. However, histological studies have shown that brain tissue taken from neurologically healthy aged individuals contains low concentrations of tau 蛋白质 having phosphate binding at four to five times the normal level. The hyperphosphorylated tau 蛋白质 weaken the integrity of the microtubule, increase the risk of neurodegeneration, and lead to the formation of neurofibrillary tangles.
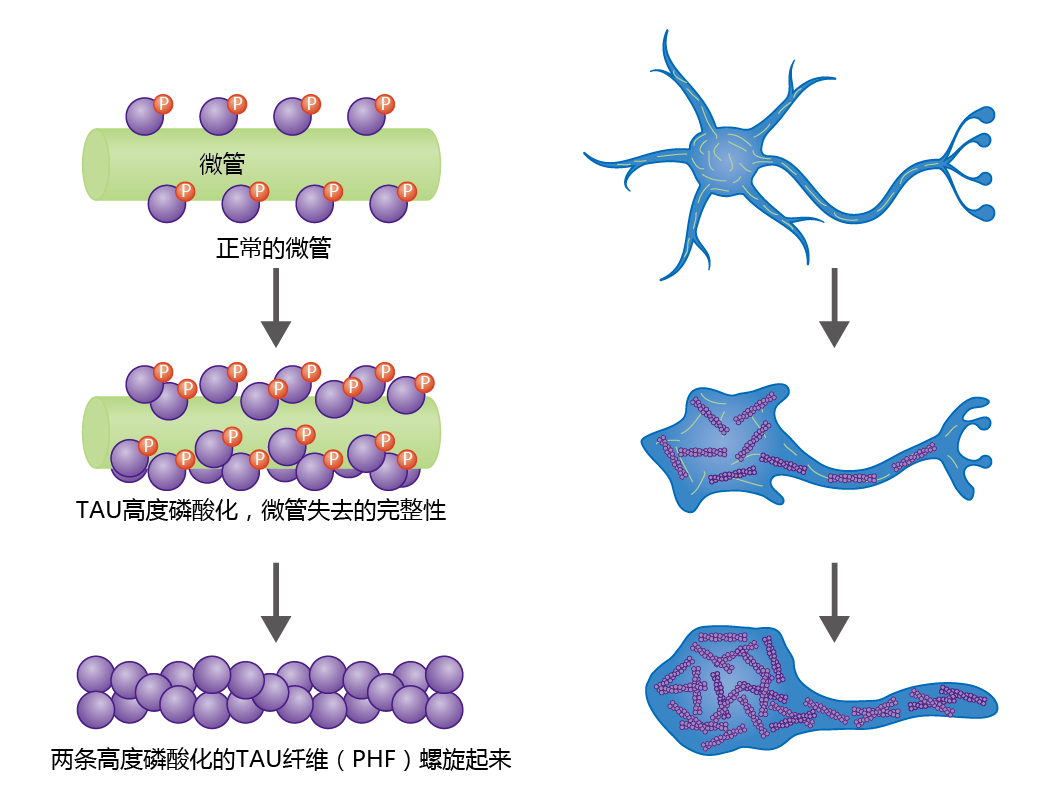
Figure 9.11 Formation of paired helical fibrils that result in neural fibrillary tangles and neural degeneration. The microtubules of the cytoskeleton maintain their integrity by means of microtubule-associated 蛋白质 (MAPs), such as the tau 蛋白质 shown here. Tau 蛋白质 bind to the microtubule surface and perform their function through phosphorylation and dephosphorylation by kinases and phosphatases. If the tau 蛋白质 become hyperphosphorylated, the microtubules lose their integrity, leading to neural degeneration. Hyperphosphorylated tau 蛋白质 aggregate into insoluble structures called paired helical fibrils (PHFs), which are the primary component of neurofibrillary tangles.
Microscopic observation shows that hyperphosphorylated tau 蛋白质 aggregates to form paired helical fibrils (PHFs), which aggregate further, leading to a breakdown in microtubule structure and disruption of intracellular transport. In turn, organelles important in maintaining the integrity of the neuron begin to break down, causing cell degeneration and dysfunction. Ultimately, PHFs replace the microtubule network, resulting in complete neural cell degeneration.
Paired helical fibrils are highly insoluble and cannot be degraded by the microglia, and thus they remain permanently affixed to the brain's extracellular matrix. Controversy exists about the effect of tangles on brain function. One theory suggests that tangles are inert and simply represent the end process of age-related neural degradation. Others suggest that tangles have a toxic effect on healthy nerve cells, which, in turn, induces further formation of PHFs.
9.2.3 阿兹海默症 is an age-related, nonreversible brain disorder
Alzheimer's disease, named after Alois Alzheimer (1864–1915), is a type of age-related dementia that causes problems with memory, thinking, and behavior. There are three primary types of Alzheimer's: early-onset, late-onset, and familial. Early-onset is a rare form of Alzheimer's in which people are diagnosed with the disease before age 65. Most cases occur in 40- to 50-year-old patients with Down's syndrome, an observation implicating the involvement of genes on Chromosome 21. Early-onset Alzheimer's progresses more rapidly than the other two forms and leads to brain abnormalities not observed in late-onset Alzheimer's disease. The late-onset disease, the most prevalent form (80–90% of all Alzheimer's cases), normally begins after the age of 65(TABLE 9.2). Whether late-onset Alzheimer's disease has a genetic component remains unknown. Most epidemiological surveys have not been able to show a significant family history for the lateonset disease. In contrast, familial Alzheimer‘s disease (FAD) has a direct genetic component; it accounts for less than 5% of Alzheimer's cases. This form normally affects individuals in their early forties. The genes associated with FAD appear to be located on three chromosomes: 1, 14, and 21.
The principal features of all three types of Alzheimer's disease are the same and are widely described, but the cause appears to be multifactorial, varies with the type of disease, and remains unknown. As noted above, mutations in genes on Chromosomes 1, 14, and 21 have been shown to be linked to Alzheimer's disease, but they seem to directly affect only those with FAD. The mutations associated with FAD all have a role in the processing of APP and may lead to the accumulation of amyloid plaques. Neither genetics nor family history has been strongly correlated with late-onset Alzheimer‘s. Nonetheless, a polymorphism, an allele variant, on Chromosome 19 has been identified as increasing the risk of developing Alzheimer's. Inflammation, oxidative stress, and disruption in the production of neurotransmitters have also been suggested as possible causes of Alzheimer's disease. The evidence sup- porting a direct link between these potential causes and Alzheimer's is not yet strong enough to warrant inclusion here.
9.2.4 阿兹海默症 begins in the entorhinal cortex and progresses into the cortex
All types of Alzheimer's disease show a similar progression of symptoms and brain pathology (Figure 9.12) . The early or pre-clinical pathology begins in the entorhinal cortex, a small structure at the base of the hippocampus that is responsible for relaying information between the cortex and hippocampus. Some estimates suggest that pre-clinical pathology can begin 10–15 years before symptoms appear. The mild loss of function in the hippocampus during the preclinical stage leads to some memory loss, a symptom that may be noticed only in retrospect, after diagnosis. As the disease progresses into the mild or clinical stage, cortical shrinkage occurs in the frontal, temporal, and occipital lobes. The symptoms that begin during this stage reflect effects in areas of the cortex (see Table 9.1). That is, loss of neurons in the cortical areas results in declining language skills (temporal lobe), mild loss in reasoning (frontal lobe), and hallucinations (occipital lobe). The patient also experiences changes in circadian rhythm that may lead to day-night reversal, such as sleeping during the day and being awake at night. The day-night reversal disappears in many Alzheimer's patients as the disease progresses to more advanced stages. The patient's sense of personality and relationship to the world also begins to deteriorate during the mild stage. Amyloid plaques and neurofibrillary tangles spread into the brain stem, affecting autonomic function. In particular, the locus coeruleus, located in the pons, becomes affected, causing a decline in the ability to regulate stress and panic.
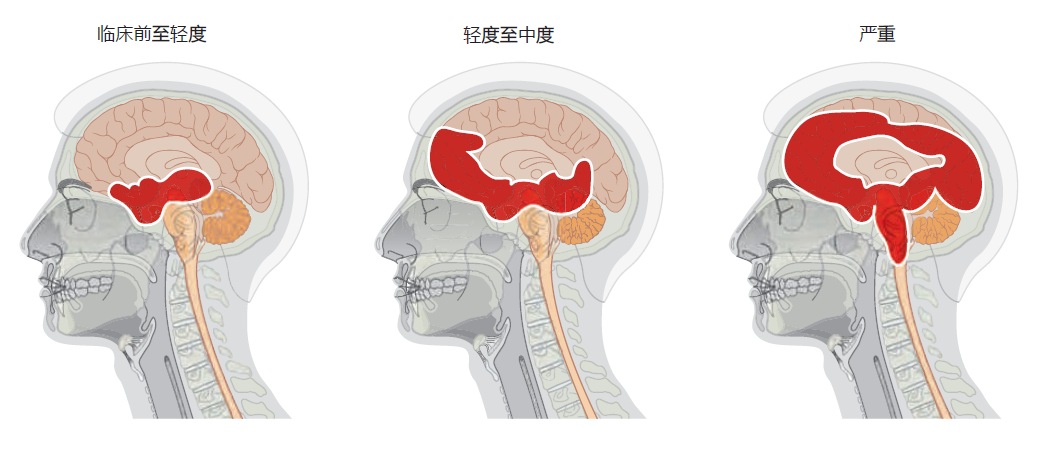
Figure 9.12 Progression of Alzheimer's disease from preclinical to severe. Alzheimer's begins subtly, and without symptoms, in the entorhinal cortex, which lies just below the hippocampus. With the transition from mild to moderate Alzheimer's disease, the hippocampus shrinks considerably and neurofibrillary tangles and amyloid plaques begin to appear in the cortex. Areas in the upper brain stem are also affected during the moderate stage. In the final stage of Alzheimer's, large areas of the cortex are involved. The disease spreads into the autonomic centers of the medulla oblongata, causing difficulties in respiration. Death due to pneumonia or heart failure is common.
In the final stage of Alzheimer's disease, also referred to as the demented stage, the hippocampus is half the size of that found in a normal brain, resulting in complete loss of memory. Severe neurodegeneration in the cortex leads to an inability to speak or understand any form of communication. The patient loses all sense of self. The cerebellum atrophies, and the patient can no longer control his or her movements, resulting in confinement to bed. In the 2–3 months prior to death, the pathology spreads into the medulla oblongata, affecting basic autonomic functions such as bowel and bladder control, heart rate, swallowing, and respiration. The patient's inability to breathe properly and expel material from the lungs often leads to pneumonia and death.
9.2.5 The ε4 allele of the apolipo蛋白质 E gene is a risk因子for late-onset 阿兹海默症
Although the cause of Alzheimer's disease remains unknown, a genetic variant of the lipid-binding 蛋白质 apolipo蛋白质 E (ApoE) has been found to be more frequent in patients with late-onset Alzheimer's. The APOE gene resides on Chromosome 19 and has three common alleles, ε2 (protective against Alzheimer's), ε3 (the most frequent in Caucasians), and ε4 (a risk factor for Alzheimer's). Synthesis of ApoE occurs in the liver, and its function has been primarily associated with the transport of cholesterol in the blood (the importance of this to cardiovascular health is discussed later in the chapter). In the brain, ApoE is synthesized by astrocytes and microglia, and here it has a major role as an extracellular lipid carrier and in the maintenance of the microvasculature.
Approximately 40–60% of individuals who have at least one copy of the ε4 allele develop late-onset Alzheimer's disease. That is, having the ε4 allele does not cause Alzheimer's disease, it only increases the risk. The risk of developing Alzheimer's increases with the number of copies of the ε4 allele. Receiving only one copy increases the risk by 30% compared with those without this allele variant. A person who receives a copy of the ε4 allele from both parents has a 50– 60% chance of developing the disease. These risk factors are striking when compared with the 9% lifetime risk for Alzheimer‘s disease in people without the ε4 allele. The biochemical mechanism underlying ApoE‘s impact on Alzheimer's remains, by and large, a mystery.
9.2.6 Treatments for 阿兹海默症 target neurotransmission and the prevention and degradation of amyloid plaques
In general, medications currently in use for treating patients with Alzheimer's focus on helping to delay symptoms or prevent them from becoming worse for a limited period of time. Current medications do not cure or prevent the progression of the disease. The medications currently prescribed for mild to moderate Alzheimer's are known as cholinesterase inhibitors and most likely work by inhibiting the breakdown of acetylcholine, the neurotransmitter important for memory and cognition. The three most widely prescribed cholinesterase inhibitors are galantamine (Razadyne), rivastigmine (Exelon), and donepezil (Aricept ® )). Because Alzheimer's is characterized by a progressive loss in the brain's ability to synthesize acetylcholine, cholinesterase inhibitors are effective for just a limited time. The medication used for the treatment of moderate to severe Alzheimer's is memantine (Namenda ® ). The precise reason that memantine helps Alzheimer's patients think more clearly remains obscure, but the chemical is known to prevent accumulation of glutamate in the synaptic cleft. Glutamate, a neurotransmitter normally found in areas of the brain associated with cognition, can be toxic to neurons in abnormally high amounts.
Ongoing research into new treatments and therapies for Alzheimer‘s disease has focused primarily on preventing the formation of 蛋白质 aggregates or degrading them once they have formed. One hypothesis posits that plaque formation may be associated with altered immune function. This hypothesis was tested by injecting antibodies to Aβ protein into transgenic mice that overexpress Aβ 蛋白质 and accumulate amyloid plaques. Remarkably, the plaque concentration decreased significantly in mice receiving the antibodies compared with those receiving a placebo injection. Clinical trials in humans using a similar approach are currently underway.
Some of the most recent research has linked two important questions concerning the development of Alzheimer‘s disease: (1) Why do all aging brains show some accumulation of amyloid plaques, but only a small percentage experience the widespread accumulation associated with Alzheimer‘s? (2) What is the mechanism underlying the increased risk of Alzheimer‘s associated with the APOE ε4 (APOE4) gene variant? The answers to these questions may lie in an abnormal disruption in the blood-brain barrier. Recall that the blood-brain barrier protects the brain by preventing the entry of large molecules. We also know that the most common APOE variant, APOE2, expresses a 蛋白质 that supports vessel integrity, whereas the 蛋白质 expressed by the APOE4 variant seems to injure capillary cells. Using this knowledge, researchers found that transgenic mice overexpressing APOE4 showed increased damage to cells of the tight junctions that form the blood-brain barrier when compared with APOE2 transgenic mice. The resulting damage would cause holes in the tight junctions and allow large molecules in the blood, such as the Aβ 蛋白质, to breach the blood-brain barrier. If correct, these findings would help to solve the mystery of why excess amyloid plaque accumulation occurs in Alzheimer's disease but not in normal aging. Most important, this research found that the damage caused by the 蛋白质 product of APOE4 responded to pharmacological treatment in the transgenic mice, suggesting that similar treatments could be developed for humans.
9.2.7 帕金森症 is associated with loss of dopaminergic neurons
Parkinson‘s disease is an age-related, motor system disorder and usually affects people over the age of 50. The primary symptoms of Parkinson‘s—tremor or trembling of the hands, arms, legs, jaw, and face; bradykinesia, or slowness of movement; muscular rigidity; and postural instability—reflect the loss of dopamine-producing neurons in the substantia nigra region of the basal ganglia (Figure 9.13) . Patients often experience weakness and tremor on one side of the body only. Moreover, some individuals notice a “feeling of tremor” within the deep portions of large muscle groups. Tremor becomes greater during times of strong emotions, such as sexual arousal or anxiety, but subsides after the emotion has passed and returns to its normal level. An inability to create facial expressions that match the person‘s emotional state (smile, frown, and so on.) begins to appear during the early stages of Parkinson‘s disease, as does a slight change in the voice. Both effects are caused by rigidity and slowing of muscles in the head and neck. A slight stooped posture occurring during the later stage of early Parkinson's results in a feeling of being out of balance and creates difficulty in walking.
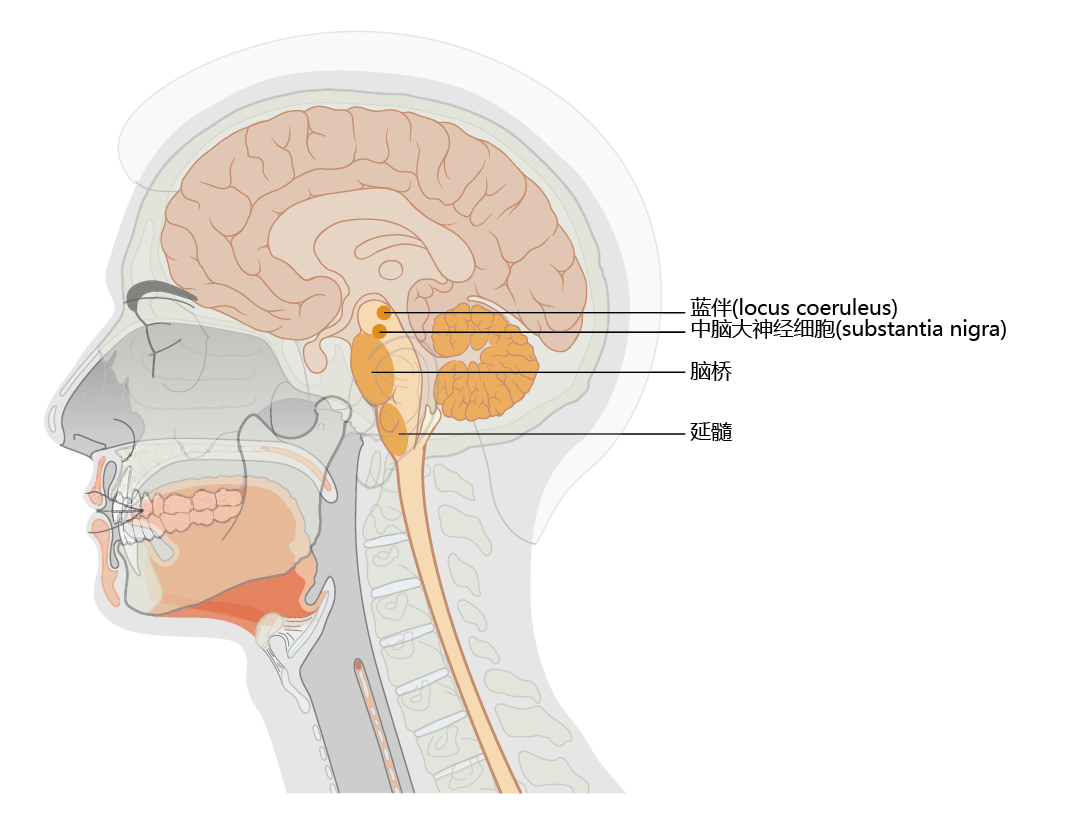
Figure 9.13 Location of the substantia nigra and locus coeruleus. Note the close proximity of the locus coeruleus to the substantia nigra. Declining synthesis of the neurotransmitter dopamine in the substantia nigra results in the early symptoms of Parkinson‘s disease. Loss of norepinephrine-producing neurons in the locus coeruleus occurs in Alzheimer‘s disease. Many Alzheimer‘s patients develop Parkinson‘s disease, and many Parkinson‘s patients develop Alzheimer‘s disease.
As Parkinson‘s disease progresses to the moderate stage, the motor function impairments become more severe. The muscle rigidity and cramps that accompany loss of motor function can cause considerable pain. While many of the effects caused by the declining ability of neurons in the substantia nigra to synthesize dopamine can be significantly reduced through drug therapy, the autonomic centers in the brain stem that synthesize acetylcholine and/or norepinephrine begin to show dysfunction. Parkinson's patients often suffer from constipation, overproduction of saliva (drooling), lack of bladder control, and an inability to regulate body temperature. Finally, some individuals might suffer from what clinicians call day-night reversal. That is, the patient is awake during the night and sleeps during the day.
Although large-scale epidemiological investigations of Parkinson's disease have not been completed, estimates from several small population studies indicate that the prevalence of Parkinson's worldwide increases after age 50. Indeed, age remains the only widely accepted risk factor for Parkinson's. Occurrence of the disease in people under 50 years of age reflects a rare genetic/family form of Parkinson's that is caused by mutations in genes found primarily on Chromosomes 1, 4, and 6. Controversy exists as to the variation of disease prevalence with respect to ethnicity/race and gender. For example, African Americans seem to have the highest rate of Parkinson's disease in the United States. However, the prevalence of Parkinson's in Nigeria is among the lowest in the world, suggesting that factors other than race account for the high rate of Parkinson's in African Americans. No clear difference has been shown in the prevalence of Parkinson's in men and women of similar age.
9.2.8 Increasing the brain's多巴胺浓度is the primary objective in treatment of 帕金森症
The cause of Parkinson's disease is loss of the neurotransmitter dopamine, and there are no treatments for reversing or curing this loss. Increasing the level of dopamine in the brain has proved effective in reducing the symptoms of Parkinson's, and there are two general approaches to this treatment. During the early stages of the disease, many physicians prescribe drugs known as dopamine agonists, which stimulate the receptors in nerves that are normally stimulated by dopamine. These drugs work only during the early stages of the disease, when there are still sufficient numbers of dopaminergic receptors/nerves to stimulate. As the disease progresses and dopaminergic receptors decline, the effectiveness of dopamine agonists declines. At this point, the focus of drug therapy shifts to increasing the concentration of dopamine in the brain. However, dopamine cannot pass through the blood-brain barrier. A precursor to dopamine, called levodopa or l-dopa, can cross the blood-brain barrier and is converted to dopamine in the brain, through a reaction catalyzed by DOPA decarboxylase. This reaction also occurs in the peripheral nervous system, however, resulting in unwanted side effects such as nausea and vomiting. In most cases, l-dopa is given together with a peripheral DOPA decarboxylase inhibitor known as carbidopa.
While the l-dopa–carbidopa combination is extremely effective at reducing the symptoms associated with Parkinson's disease, these drugs do have limits and side effects that are progressive. Among the most frequent side effects are jerky movements such as facial grimacing, postural tics, and exaggerated chewing. Long-term administration of l-dopa–carbidopa often results in low blood pressure, skin rashes, depression, and alterations in sleep patterns. The effectiveness of l -dopa–carbidopa diminishes as Parkinson's progresses and the number of dopaminergic receptors/nerves decreases.
9.2.9 Lewy bodies are the pathological hallmark of 帕金森症
The cause of Parkinson's disease—the reason for the loss of dopaminergic neurons—remains unknown, and early diagnosis is difficult, due in large part to the slow progress of the disease and clinical signs that can be confused with other neurological disorders. We have discussed how the accumulation of damaged or misfolded 蛋白质 in brain tissue is often associated with aging and age-related disease. Such 蛋白质 are also found in association with Parkinson's disease. These 蛋白质, known as Lewy bodies, are highly insoluble aggregates of fibrous proteins that appear in the cytoplasm of neurons. Although Lewy bodies can be found in aged humans without Parkinson's, their accumulation in the substantia nigra and locus coeruleus is a pathological hallmark of the disease. The major components of Lewy bodies are two 蛋白质 that normally participate in the maintenance of 蛋白质 structure, ubiquitin and α-synuclein. Ubiquitin, a small (76 amino acid) heat shock 蛋白质, attaches to misfolded or damaged 蛋白质 to mark them for degradation. The precise function of α-synuclein remains unclear, but most researchers agree that this 蛋白质 plays a vital role in the maintenance and regulation of dopamine vesicles at the synaptic terminals. Its β-sheet conformation involving specific amino acid residues also promotes the formation of β-sheet aggregates, if the 蛋白质 does not undergo degradation. These aggregates form the primary component of Lewy bodies. As in amyloid plaque accumulation in Alzheimer's disease, the accumulation of Lewy bodies in the brain in Parkinson's does not cause the disease but seems to result from it.
9.2.10 Several genes are associated with early-onset 帕金森症
Early-onset Parkinson's disease, occurring before the age of 50, accounts for less than 1% of all Parkinson's cases and may be directly linked to family history and mutations in several genes. Mutations linked to early-onset Parkinson's have been discovered at several loci, but genes associated with the 蛋白质 that constitute the majority of Lewy bodies, ubiquitin (the Parkin gene, on Chromosome 6) and α-synuclein (a gene on Chromosome 4), have drawn the most attention. The 蛋白质 product of the Parkin gene functions as a component of ubiquitin ligase, the enzyme that adds ubiquitin to other 蛋白质. A mutation in the Parkin gene disrupts the normal pathway that marks 蛋白质 for degradation, thereby allowing damaged or misfolded proteins to accumulate and 蛋白质 aggregates to form. Interestingly, the primary 蛋白质 aggregates found in Parkinson's disease, the Lewy bodies, are not found in the neurons of patients with the Parkin gene mutation. Rather, current research suggests that the mutation in the Parkin gene causes mitochondrial dysfunction and overproduction of reactive oxygen species, leading to cell death.
Regardless of the type of mutation, in the Parkin gene or in the α-synuclein gene, only about 50% of cases of early-onset Parkinson‘s disease can be linked to these mutations. Moreover, people with the late-onset disease, the type accounting for 99% of all cases of Parkinson‘s, rarely have this mutation.
9.2.11 Several factors may predispose individuals to 帕金森症
Parkinson‘s disease, as we‘ve seen, does not have a clear and unequivocal cause. It is a multifactorial disorder involving both genetic and environmental factors. Mutations of genes leading to early-onset Parkinson‘s strongly suggest a genetic component. The first clue that the environment may contribute to the disease came when drug users, in an attempt to synthesize the morphine-like drug meperidine, instead produced a neurotoxin, methyl-phenyl-tetrahydropyridine (MPTP). MPTP selectively kills dopaminergic neurons in the substantia nigra and other parts of the brain stem. Thus, when these drug users took what they believed to be meperidine, they developed an irreversible Parkinson‘s-like condition. Subsequent studies investigating the connection between MPTP and Parkinson‘s suggested that this compound inhibits a biochemical pathway in the mitochondria that protects neurons from oxidative damage. While the association between oxidative damage and Parkinson‘s remains only correlative, exposure to other agents that increase oxidative stress, such as Paraquat or high levels of iron and manganese, has been shown to increase the risk of developing Parkinson's.