
第一节 细胞形态结构的观察方法
肉眼的分辨率一般只有0.2mm,很难识别单个细胞;光学显微镜的分辨率可达0.2μm,借此发现了细胞;电子显微镜分辨率高达0.2nm,将细胞的超微结构展现在人们面前。图2-1比较了光学显微镜、电子显微镜和扫描隧道显微镜适宜观察的样品大小及分辨率。有趣的是,光学显微镜、电子显微镜和扫描隧道显微镜,除了它们因提高分辨率而称之为显微镜这一共性外,在其成像原理、仪器构造以及使用和操作方法等方面,几乎没有任何共同之处。

一、光学显微镜
17世纪,光学显微镜(light microscope)的发明使人们第一次看见了细胞,进而建立了细胞学说,为细胞学的兴起和发展打下基础。光学显微镜至今仍然是细胞生物学研究的重要工具。随着光学显微术与图像处理技术的快速发展,光学显微镜在研究细胞的结构与功能,特别是生物大分子在活细胞中的定位及其动态变化和相互作用等方面展示出了新的活力。
(一)普通复式光学显微镜
光学显微镜主要由三部分组成:①光学放大系统(图2-2),为两组玻璃透镜:目镜与物镜。②照明系统:包括光源和聚光镜,有时另加各种滤光片以控制光的波长范围。③镜架及样品调节系统。1872年,德国科学家阿贝(E.K.Abbe)等研制出了完美程度几乎可与现代普通光学显微镜媲美的复式显微镜。
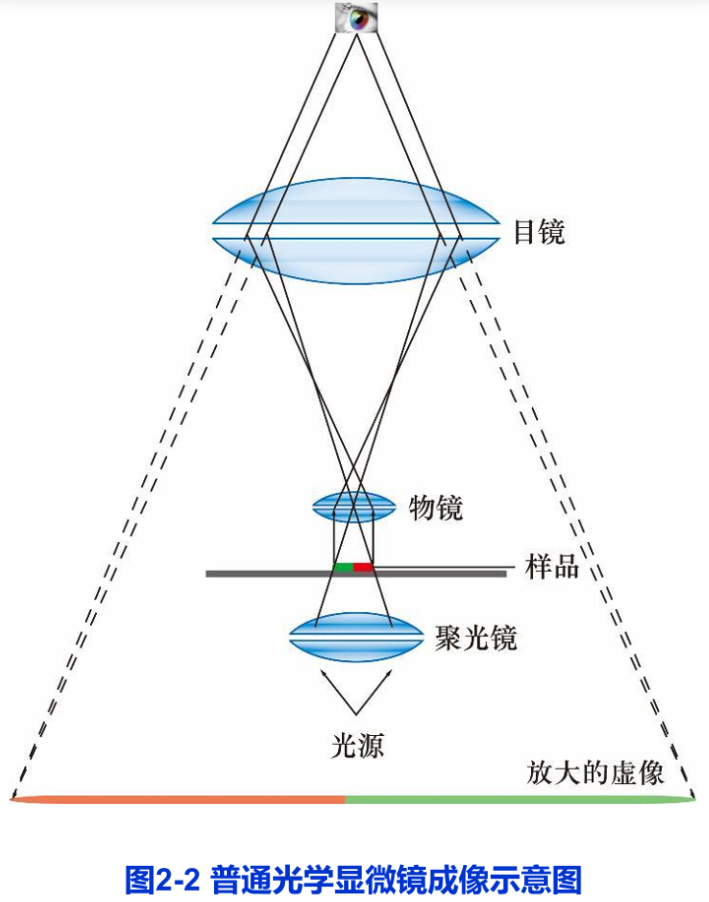
显微镜最重要的性能参数是分辨率(resolution),而不是放大倍数。分辨率是指能区分开两个质点间的最小距离。分辨率D的高低取决于光源的波长λ,物镜镜口角α(标本在光轴上的一点对物镜镜口的张角)和介质折射率N(图2-3),它们之间的关系是:
D=(0.61λ)/N·sin(α/2)
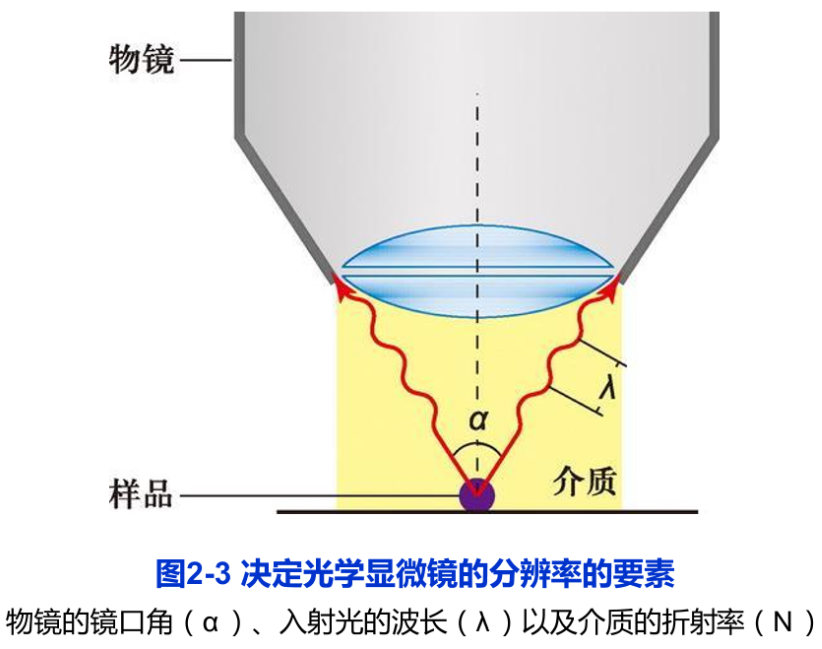
通常a最大值可达140°,空气中N=1,最短的可见光波长=400nm,此时分辨率D=260nm,约0.3μm。若在油镜下,N可提高到1.5,分辨率D则可达02m,所以普通光学显微镜的最大分辨率是0.2μm。
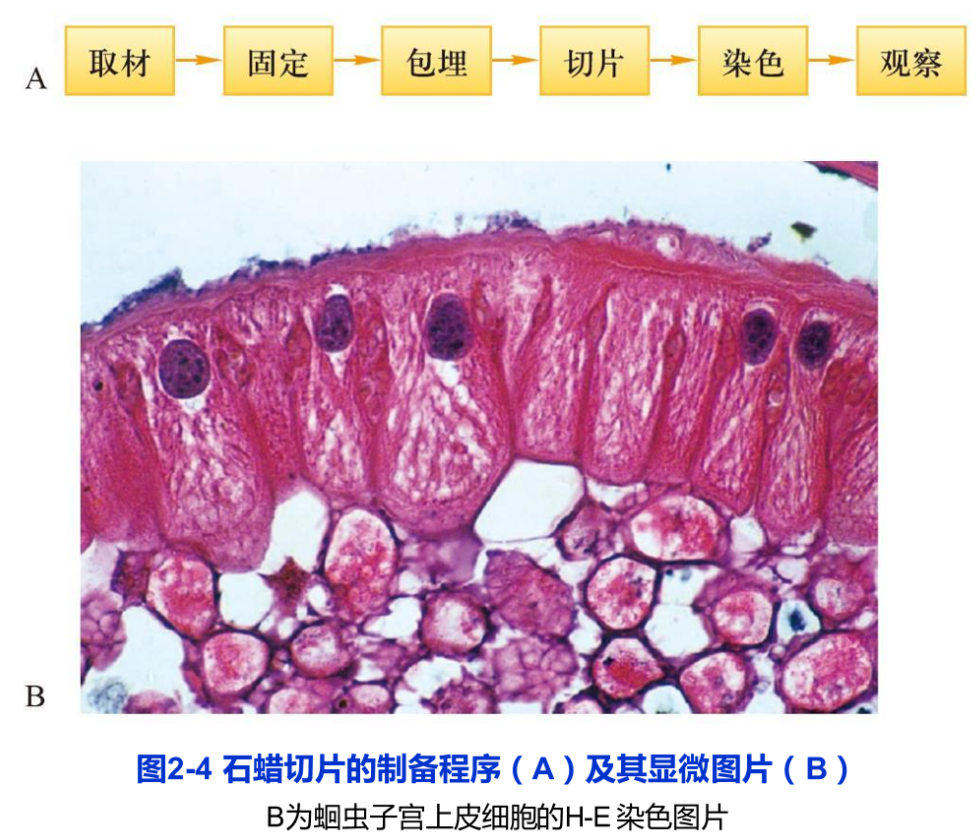
光学显微镜可以直接用于观察单细胞生物或体外培养细胞。如果观察生物组织样品,则通常需要对所观察的材料进行固定和包埋(常用的固定剂如甲醛,包埋剂如石蜡等),再将包埋好的样品切成厚度约5μm的切片,最后进行染色(图2-4A)。如苏木精和伊红染色(又称HE染色)就是一种常用的染色方法。碱性染料苏木精和酸性染料伊红分别与细胞核和细胞质的某些成分特异性地结合,从而改变透射光线的波长,我们就可以清晰观察到蓝紫色的细胞核和红色的细胞质的形态(图2-4B)。
(二)相差显微镜和微分干涉显微镜
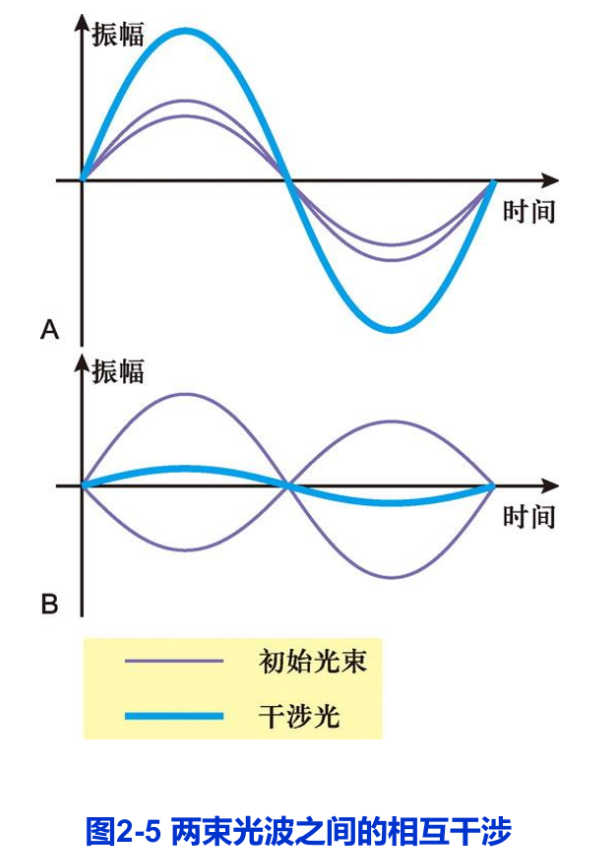
生物样品一经固定就失去了生物活性,活细胞显微结构的细节可以借助相差显微镜(phase-contrast microscope)来观察。光波的基本属性包括波长、频率振幅和相位等。可见光的波长和频率的变化表现为颜色的不同,振幅的变化表现为亮暗的区别,而相位的变化却是人眼不能觉察的。当两束光通过光学系统时,会发生相互干涉。如果它们的相位相同,干涉的结果是使光的振幅加大,亮度增强。反之,就会相互抵消而亮度变暗(图2-5)。相差显微镜和干涉差显微镜就是利用这样的原理增强样品的反差,从而实现对非染色活细胞的观察。
光线通过不同密度的物质时,其滞留程度也不同。密度大则光的滞留时间长,密度小则光的滞留时间短,因而,其光程或相位发生了不同程度的改变。相差显微镜是在普通光学显微镜的基础上,添加两个元件,即环状光阑”和在物镜后焦面上的“相差板”,从而可将这种光程差或相位差,通过光的干涉作用,转换成振幅差,因此,可以分辨出细胞中密度不同的各个区域(图2-6)。1932年,德国物理学家P. Zernike制备出第一台相差显微镜,他因此项发明在1953年获诺贝尔物理学奖。
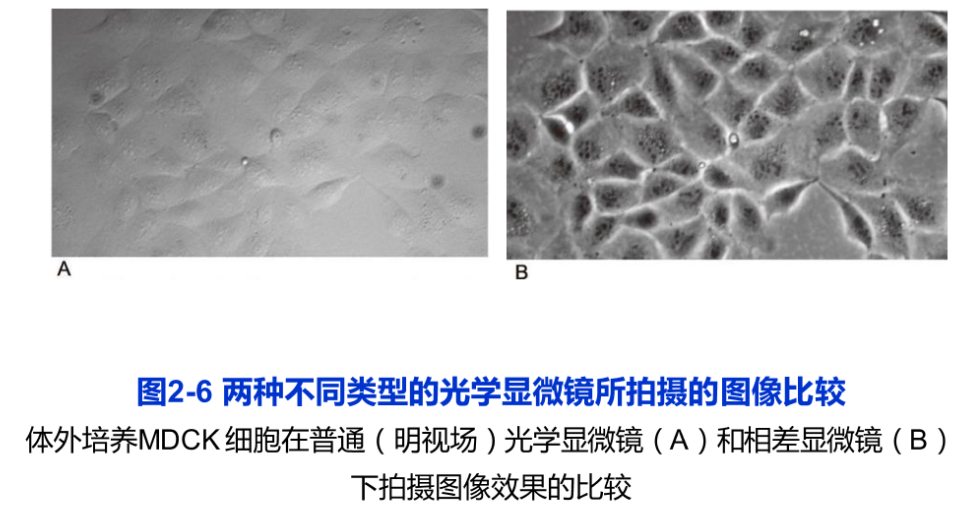
1952年,G. Nomarski在相差显微镜的基础上,发明了微分干涉显微镜 (differential-interference microscope),又称Nomarski相差显微镜。微分干涉显微镜是以平面偏振光为光源。光线经棱镜折射后分成两束,在不同时间经过样品的相邻部位,然后再经过另一棱镜将这两束光汇合,从而使样品中厚度上的微小区别转化成明暗区别。微分干涉显微镜更适于研究活细胞。
如果将微分干涉显微镜接上高分辨率录像装置,就可以用来观察并记录活细胞中的颗粒及细胞器的运动。计算机辅助的微分干涉显微镜可以进一步提高样品的反差并降低图像的背景“噪声”,使一些常规光学显微镜难于观察的一些显微结构,如单根微管(直径约24nm)等也可以在光镜下分辨出来。其分辨率比普通光镜提高了一个数量级。从而为在高分辨率条件下研究活细胞提供了必要的工具。应用这一原理制备的录像增差显微镜(video-enhance microscope)可以用来直接观察颗粒物质沿着微管运输的动态过程。
(三)荧光显微镜
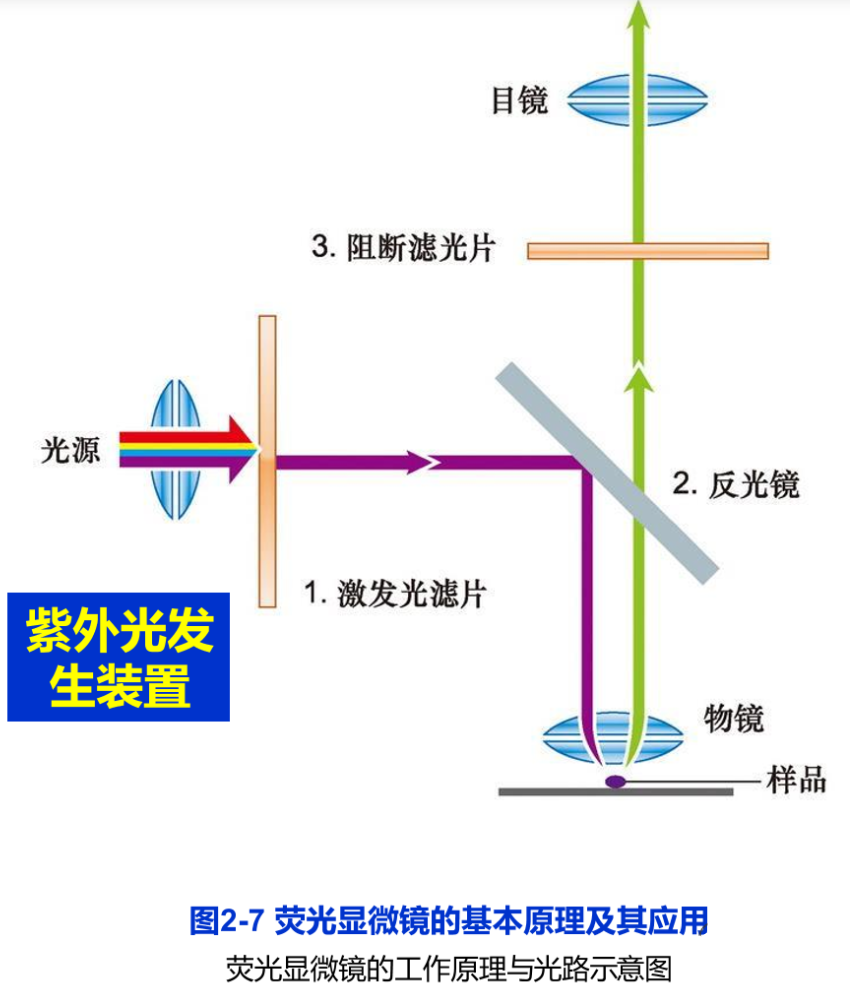
荧光显微镜(fluorescence microscope)是在光镜水平上,对细胞内特异的蛋白质、核酸、糖类、脂质以及某些离子等组分进行定性定位研究的有力工具。荧光显微镜采用的光源和光学系统均与普通光学显微镜有所不同,其核心部件是滤光片系统以及专用的物镜镜头(图2-7A)。滤光片系统由激发滤光片(安装在光源和样品之间,只允许特定波长的激发光通过)和阻断滤光片(安装在物镜和目镜之间,只容许荧光染料所发出的荧光通过)组成。这样,通过激发滤光片的短波长的激发光,照射标记在样品中的荧光分子上,使之产生一定波长的可见光即荧光(图2-7B)。由于荧光显微镜的暗视野为荧光信号提供了强反差背景,非常微弱的荧光信号亦可得以分辨。
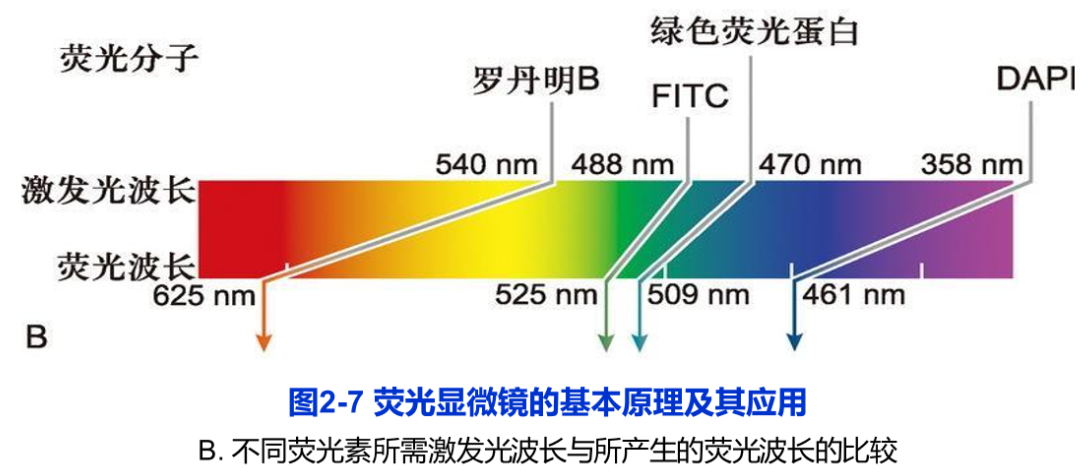
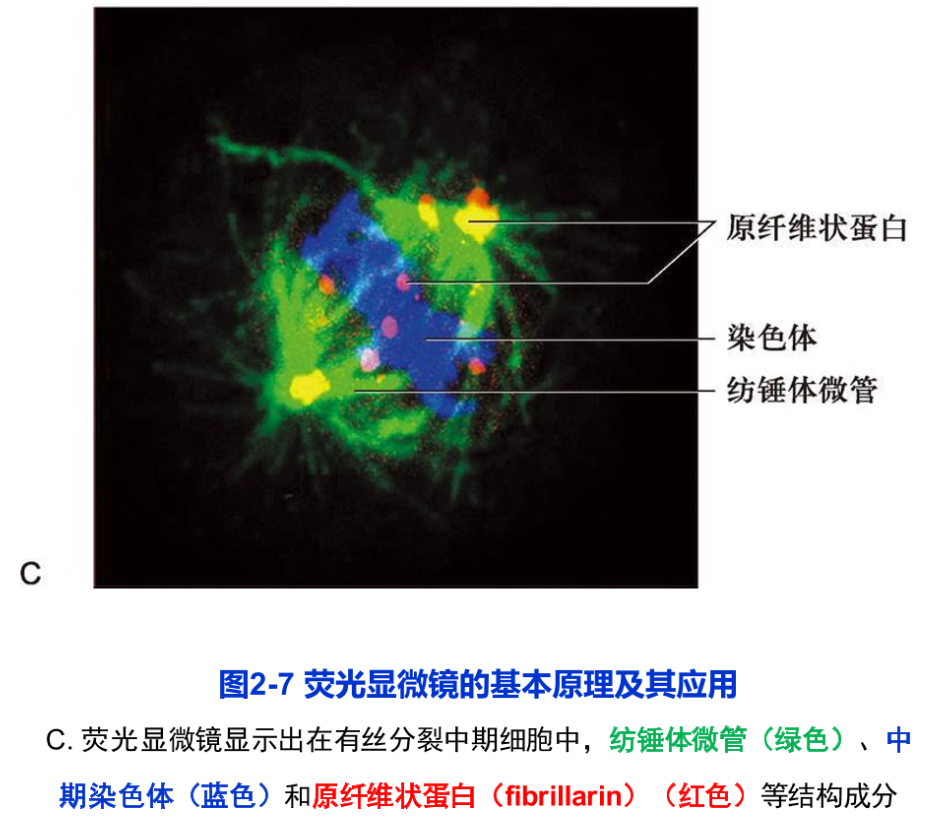
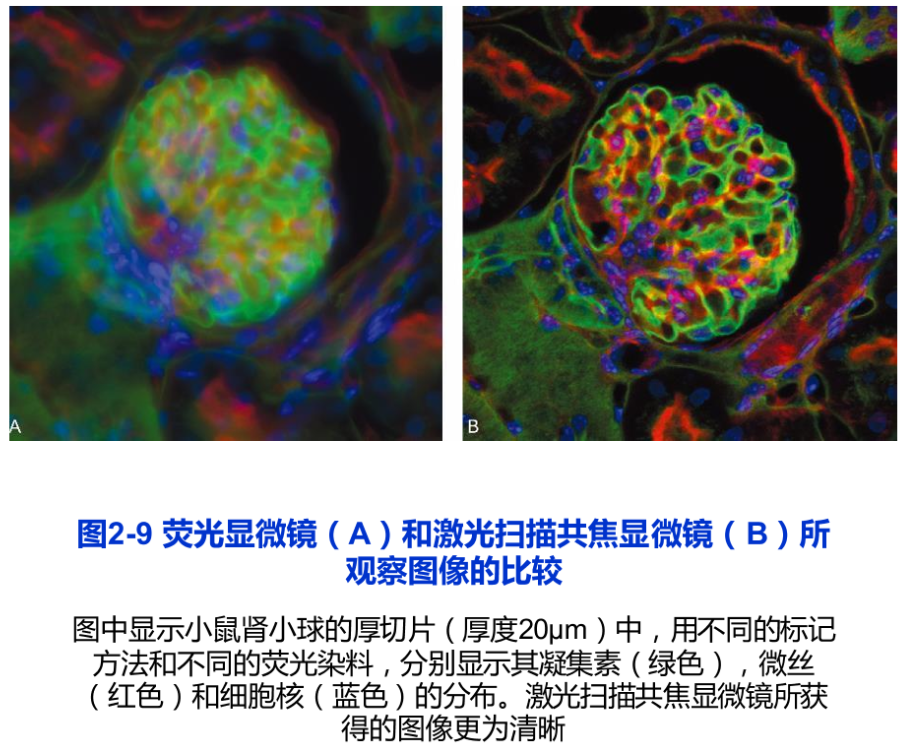
荧光显微镜样品制备技术包括免疫荧光技术和荧光素直接标记技术。荧光染料DAPI特异性地直接与细胞中的DNA相结合,从而显示出细胞核或染色体在细胞中的定位。这是一种常用的直接标记技术。目前,荧光显微术得到了广泛的应用,特别是细胞内动态变化的研究。例如,用显微注射器将标记荧光素的肌动蛋白注射到培养细胞中,可以看到肌动蛋白分子组装成肌动蛋白纤维的过程。又如,将在激发光作用下,可产生荧光的绿色荧光蛋白(green fluorescent protein,gP)的基因与编码某种蛋白质的基因相融合,利用荧光显微镜,就可以在表达这种融合蛋白基因的活细胞中,观察到该蛋白质的动态变化。由于不同荧光素所激发出的荧光波长不同,同一样品可以用两种以上的荧光素标记,从而同时显示不同成分在细胞中的定位(图2-7C,图2-9A)。
(四)激光扫描共焦显微镜
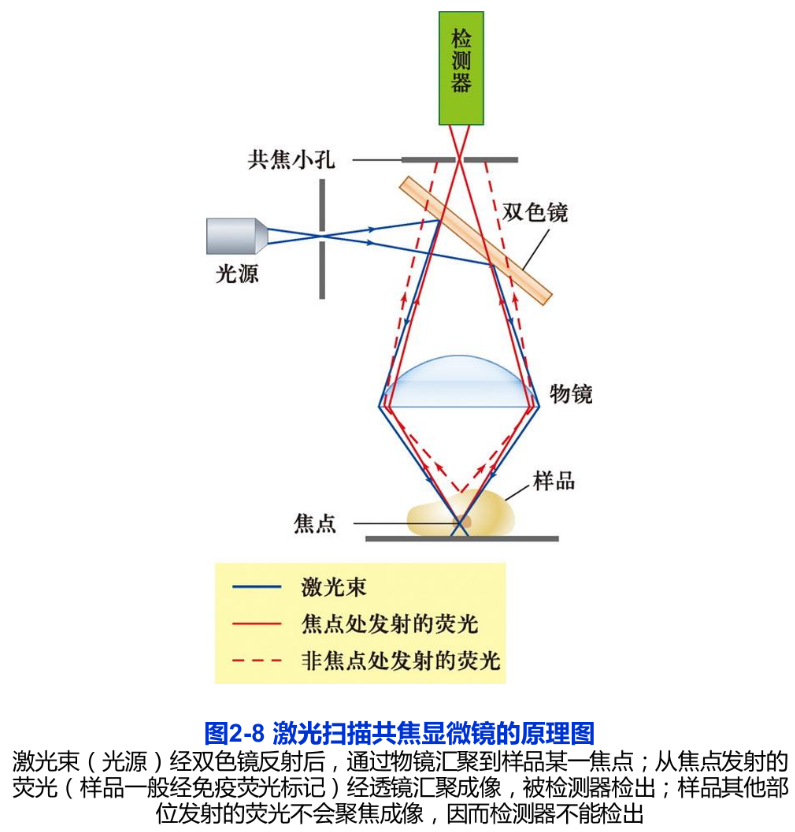
在普通荧光显微镜下,许多来自样品中焦平面以外的荧光会使观察到的图像反差减小、分辨能力降低。而激光扫描共焦显微镜(laser scanning confocal microscope, LSCM 简称共焦显微镜)相当于在荧光显微镜上安装了一套激光共焦成像系统,并以激光(可见或紫外激光)为光源,从而,极大地提高了图像的分辨率(图2-8)。所谓共焦,是指聚光镜和物镜同时聚焦到同一点上,因而只有聚光镜聚焦在样品中的某一位点上所产生的激发荧光才能清晰成像,而来自焦平面以外的散射光则被小孔或狭缝挡住,再利用激光扫描装置和计算机高速采集与处理会聚每一个点上的信息,形成一幅清晰的二维图像。激光扫描共焦显微镜的分辨率可以比普通荧光显微镜的分辨率提高1.4~1.7倍。其纵向分辨率(axial resolution)也得到很大的改善。由于可自动调节并改变观察的焦平面,所以可以通过“光学切片”即改变焦点获得一系列不同切面上的细胞图像,经叠加后重构出样品的三维结构。早在1955年,M. Minsky就发明了扫描共焦显微镜。但是直到30年后,随着激光光源的应用和一系列的改进,这种新型的显微镜才得以问世。目前,它在研究亚细胞结构与组分的定位及动态变化等方面的应用越来越广泛,其中包括本章后面所谈到的荧光共振能量转移技术、荧光漂白恢复技术等,都离不开激光扫描共焦显微镜(图2-9B)。
(五)超高分辨率显微术
根据传统的光学理论,远场光学显微镜在x、y轴向的理论分辨率的极限值为200nm,而z轴向的分辨率还要比这个数值低得多,大约是500~800nm,远远不能满足要求。于是,人们开始寻找突破光学显微镜分辨率极限的方法。经过20多年的努力,研制出诸如 TIREM、PALM/ STORM、4π和STED显微术,以及SIM等多种不同类型的超高分辨率显微术。
1.全内反射荧光显微术(total internal reflection fluorescence microscopy, TIRFM)
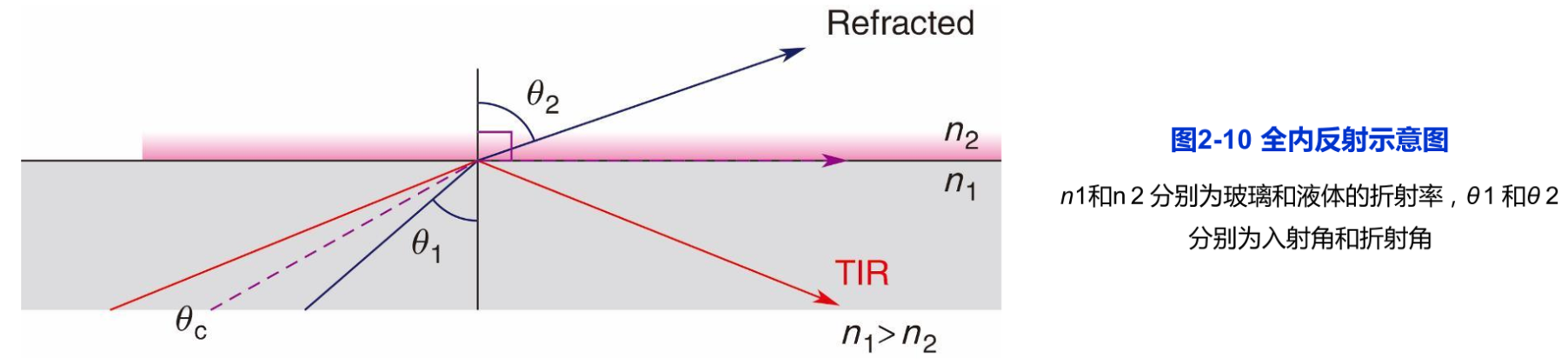
根据瑞利/阿贝公式,显微镜的分辨率主要取决于光线的波长和物镜的数值孔径。而且,由于成像过程中产生的泊松噪声和检测元件的背景噪声使光学显微镜的实际分辨率远远低于理论值。可见,降低噪声是提高分辨率的有效途径。激光扫描共焦显微镜主要是依靠针孔来减少非焦平面的信息从而提高图像的清晰度,但针孔不能无限缩小。而全内反射荧光显微镜的原理是基于斯涅耳定律,即当光线从光密介质进入光疏介质时,部分光会发生折射,而另一部分光线会发生反射(图2-10)。并且,当入射角(θ1)的角度增大时,折射角(θ2)也随之增大。当入射角达到临界角(θc)时,折射角的角度达到90°。当入射角继续增大时,就会发生全内反射(TIR)现象。此时,光线会在介质的另一面产生隐失波。隐失波的能量范围通常在200nm以内,且在z轴上呈现指数衰减,因此可以激发很薄一层(约100nm)样品内特定的荧光分子发光,这样就大大降低了背景噪声的干扰,提高了图像分辨率。该技术的特点是只能观察细胞紧靠玻片的大约100nm的范围,例如观察细胞质膜以及紧靠细胞质膜的细胞骨架组分的动态行为,而对细胞的其他部分的观察则无能为力。
2.基于单分子成像技术的PALM/ STORM方法
对于单个荧光光源的光子分布,通过特定的数学方法(如高斯函数)拟合,可以比较容易地定位荧光光源,精度达到纳米量级。P. Selvin研究小组应用单分子成像技术在体外条件下分析肌球蛋白分子的行为,达到1.5nm的定位精度。但是,如果两个荧光光源相距很近并且同时发光,应用该技术并不能区分它们。
一种绿色荧光蛋白的突变体(PA-GFP)的出现使情况出现了转机。PA-GFP在激活之前对488nm的光没有反应,但如果用405nm的激光激活一段时间,再用488nm激光照射时,可发出绿色荧光。E. Betzig等将PA-GFP的发光特性与单分子荧光的定位精度相结合,成功地突破了光学显微镜的分辨率极限。其基本原理是将目的蛋白克隆到PA-GF表达载体,并转染细胞。待融合蛋白表达后用较低能量的405nm激光照射细胞,随机地使很少一部分(相隔较远的)PA-GFP激活,再用488nm激光激发激活的PA-GFP发出绿色荧光,通过高斯函数拟合将这些荧光分子定位,然后用488nm激光将这些已经精确定位的荧光分子漂白,使其不能在下一轮激光照射时被激活。重复上述过程,每次都用405nm和488nm激光来激活、激发、定位和漂白少量的荧光分子,如此持续进行数百个循环,直到将细胞内几乎所有的荧光分子都精确地定位,并将所得到的图像合成在一张图上。这样就可以得到分辨率比传统的光学显微技术高10倍以上的图像。由此建立了光激活定位显微术(photoactivated localization microscopy,PALM)后来 Betzig的研究小组进一步发展PALM,成功记录了细胞内两种蛋白质的相对位置。PALM的缺点是只能用于观察外源表达蛋白的定位,并且每一幅图像照相所需的时间很长,不适合用于活细胞动态观察。
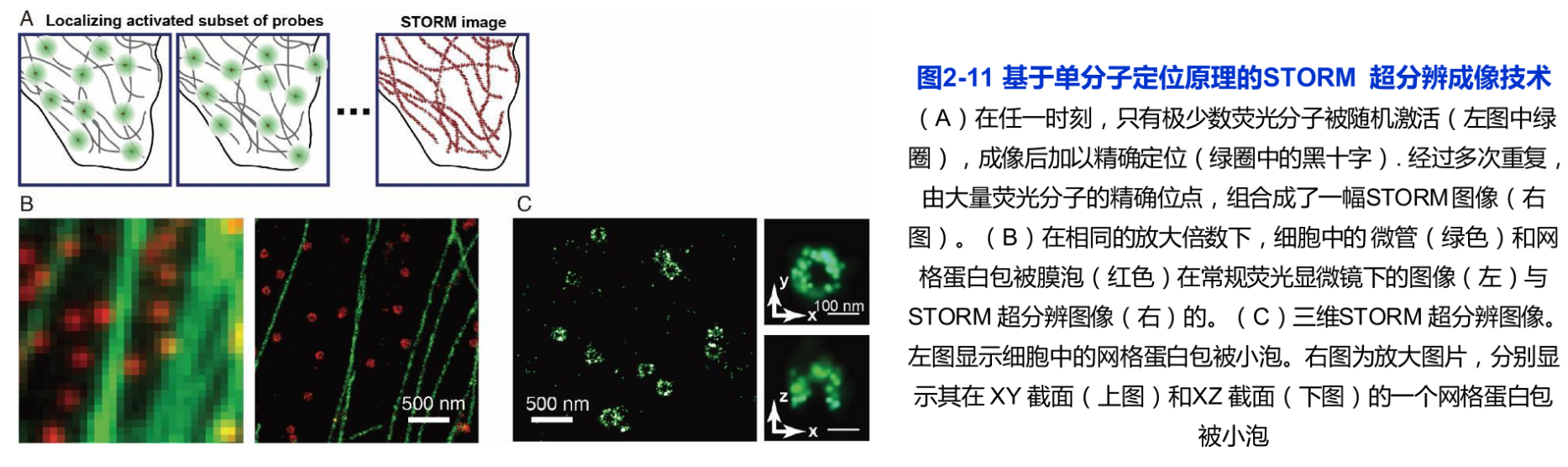
庄小威的研究小组开发了一种与PALM类似,但可以用来做细胞内源性蛋白的超分辨率定位的方法。其原理是基于花青染料可以被一种波长的光激活发出荧光,还可以被另一种波长的光波关闭而处于暗态,可以用不同波长的光波照射这些花青染料以控制它们发光或不发光。这样,就可以将原本靠得很紧的蛋白质用成对的染料(如Cy3/Cy5)标记的特异抗体反应,在显微镜下先用激光使报告分子Cy5失活,然后用561nm激光随机地激活少数几个Cy3,并导致其配对的Cy5活化,再用647nm激光激发Cy5,使其发出670nm的荧光,信号被相机捕捉后通过高斯拟合求出其中心位置。重复上述过程10000次以上,以得到100万个以上的数据,就可以重构出内源蛋白分布的高分辨率图像,该方法被命名为随机光学重构显微术(stochastic optical reconstruction microscopy, STORM)。采用不同的荧光染料对,应用 STORM可以对多达4种内源性蛋白进行精细定位,分辨率可以达到20nm左右(图2-11)。其缺点是得到一张超分辨图像的时间很长,在活细胞中很难实现。
3. 4π和STED显微术
S.ell发明的4π显微镜是基于增加物镜的接受角而等效增加物镜的数值孔径(NA)。具体是在样品的两侧各放置一个相同的高性能物镜,使总的接受角接近4π,进而提高了NA。Hell的这一设计成功地将z轴分辨率从500nm提高到100nm左右,但4π显微镜并没有突破衍射极限。此后,Hell设想使用一束激光,仅仅激发一个点的荧光基团并使其发出荧光,然后再用第二束象面包圈那样的环状高强度的脉冲激光照射那个点周围的荧光,该激光的波长可以使发光物质从受激状态返回到基态。这样,就只有中间没有完全损耗的荧光分子可以观察到。环状激光中间的孔径越小,图像的分辨率就越高。Hell给这项发明取名受激发射损耗(stimulated emission depletion,STED)显微术(图2.12)。理论上,STED显微术的分辨率不受光的衍射过程所限制,利用该技术可以使水平面的分辨率达到纳米级,而且可以快速地观察活细胞内的实时动态变化,如记录神经元突触小泡的行为,但没有提高z轴方向的分辨率。如果将4π和STED结合,将可以同时提高z轴方向的分辨率。

4.结构照明显微术
M.Gustafsson博士将非线性结构照明部件引入到传统的显微镜上,使显微镜的硬件系统增加了光栅和控制元件,软件系统可对所获取的图像信息进行计算。其原理是通过光栅的旋转和移动将多重相互衍射的光束照射到样本上,并在此发生干涉,然后从收集到的发射光模式中提取高分辨信息,生成一幅完整的图像。这一技术称为结构照明显微术(structured-illumination microscopy,SIM)。利用结构照明术可使x、y平面分辨率达到100nm,z轴向分辨率也提高了2倍。其优点是对于普通的免疫荧光标记样本和各种荧光蛋白表达样本,可以不经特殊处理直接观察。其缺点是分辨率远低于其他超高分辨率显微术(图2-13)。

二、电子显微镜
光学显微镜技术在细胞生物学研究中起着极为重要的作用。但由于光波波长的限制,光镜的分辨率难以得到进一步提高。只有借助分辨率更高的电子显微镜(electron microscope,EM,简称电镜),才可能观察到细胞内部的精细结构。电子显微镜经过了多位科学家共同努力,最终由德国科学家E. Ruska于1932年制作而成,Ruska因此于1986年获诺贝尔物理学奖。
(一)电子显微镜的基本知识
电子显微镜的基本原理与光学显微镜完全不同,构造也要比光学显微镜复杂得多。但电子显微镜的光路却与光学显微镜具有相似性(图2-14)。
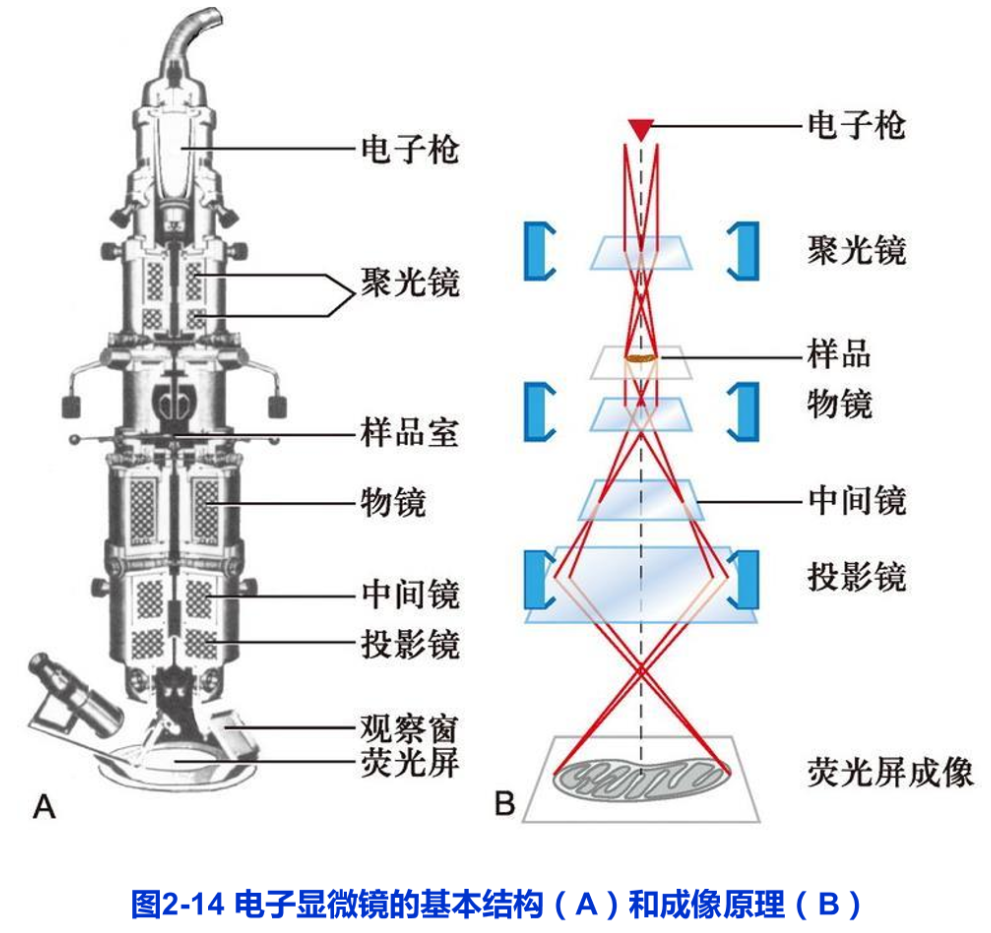
1.电子显微镜与光学显微镜的基本区别
电子显微镜的高分辨率主要是因为使用了波长比可见光短得多的电子束作为光源,波长一般小于0.1nm光源的差异决定了电镜与光镜的一系列不同点(表2-1):比如电子显微镜需要通过电磁透镜聚焦,电镜镜简中要求高度真空,图像需要通过荧光屏、感光胶片或电荷耦合器件(charge-coupled device,CCD)进行显示和记录等。
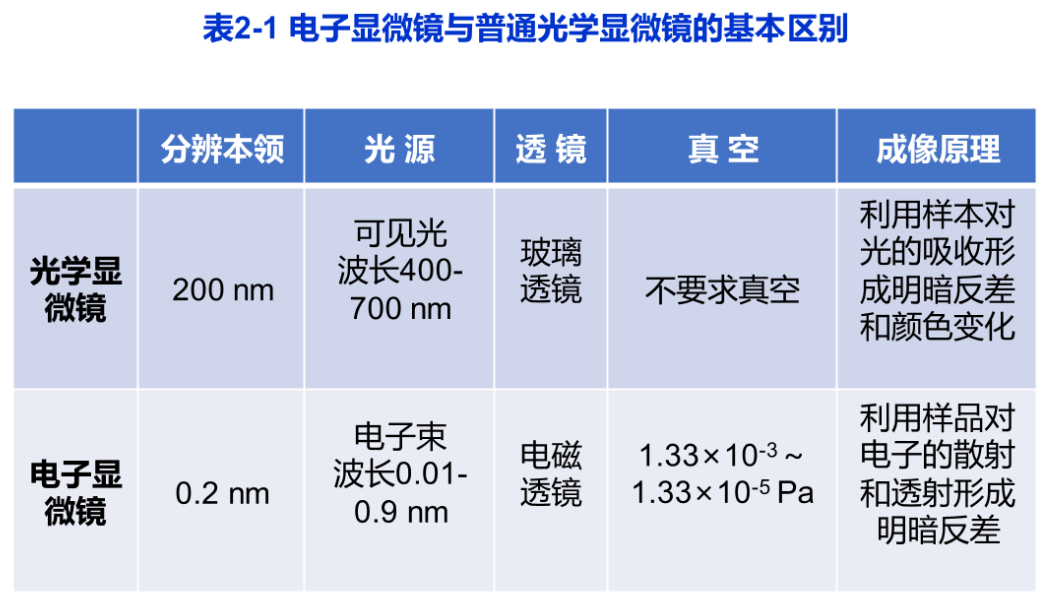
2.电子显微镜的有效放大倍数与分辨本领
如前所述,人眼的分辨率一般为0.2mm,光学显微镜的分辨率为0.2μm左右,其放大倍数为0.2mm/0.2μm,即1000倍。而电子显微镜的分辨率可达0.2nm,其放大倍数为10倍。上述放大倍数称之为有效放大倍数。如果通过光学手段继续放大,再也不会得到任何有意义的信息,因此称之为“空放大”。
电镜的分辨率与分辨本领并不等同,电镜的分辨本领是指电镜处于最佳状态下的分辨率。实际情况下,电镜的实际分辨率常常受到生物制样技术本身的限制,如在超薄切片样品中其分辨率约为超薄切片厚度的1/10,即通常超薄切片厚度若为50nm,则实际分辨率约为5nm,远低于电镜的分辨本领0.2nm。
3.电子显微镜的基本构造
电子显微镜主要由以下4部分组成(图2-14):
(1)电子束照明系统 包括电子枪和聚光镜。如可用高频电流加热钨丝发出电子,通过高电压的阳极使电子加速(这一装置称电子枪),射出的电子经聚光镜会聚成电子束。
(2)成像系统 包括物镜、中间镜与投影镜等。它们是经过精密加工的中空圆柱体,里面装置线圈,通过改变线圈的电流大小,调节圆柱体内空间的磁场强度。电子束经过磁场时发生螺旋式运动,最终的结果如同光线通过玻璃透镜时一样,聚焦成像。
(3)真空系统 用两级真空泵不断抽气,保持电子枪、镜筒及记录系统内的高度真空,以利于电子的运动。
(4)记录系统 电子成像须通过荧光屏显示用于观察,或用感光胶片或CCD相机记录下来。
(二)主要电镜制样技术
1.超薄切片技术
由于电子束的穿透能力有限,为获得较高分辨率切片厚度一般仅为40~50nm,即一个直径为20μm的细胞可切成几百片,故称超薄切片(ultrathin section)。这需要样品既要有一定刚性又要有一定韧性,而生物样品并不具备这些特性,为此,样品往往需要包埋在特殊的介质中。但包埋的过程会破坏样品的细微结构,所以超薄切片样品制备的第一步就是样品的固定,以期更好地保存细胞的精细结构(图2-15)。
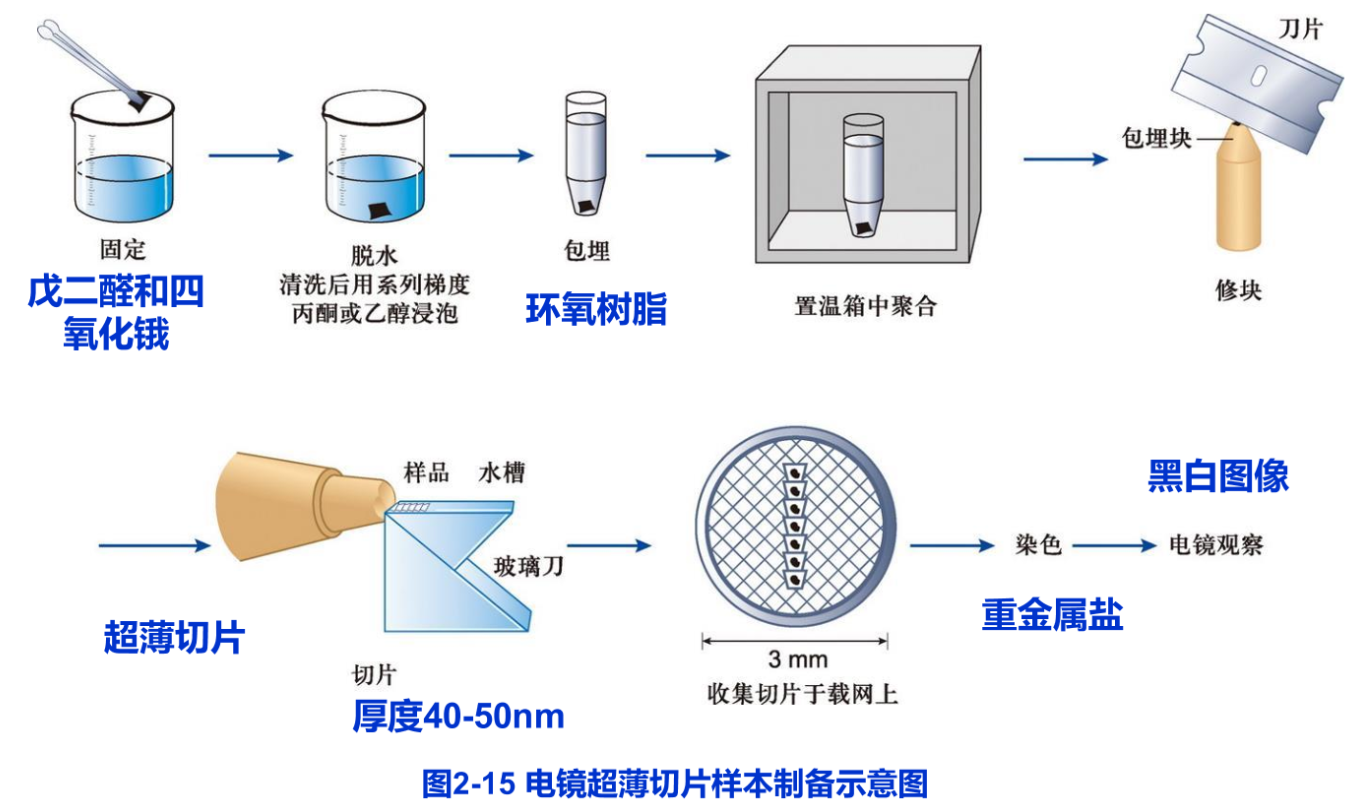
(1)固定 固定是保持样品的真实性的一个最重要的环节。固定不仅要求保持样品的形态和精细结构不发生改变;有时还要求细胞内部的成分保持在原来的位置上,甚至其免疫原性尽可能的不发生改变。透射电镜常用化学法进行样品的固定,固定剂为戊二醛和四氧化锇(OsO4,常称为锇酸)等(表2-2)。同时,为了更好地保存细胞的超微结构,还可以利用物理方法,如超低温冷冻等方法进行固定。在固定操作过程中,动物的处死和取材都要快速进行,以防细胞自溶作用造成的损伤。固定的样品块直径一般小于1mm,以便固定剂迅速渗透。
(2)包埋 包埋的目的是保证在切片过程中,包埋介质能均匀良好地支撑样品,以便获得连续、完整并有足够强度的超薄切片。包埋介质要求具有良好的机械性能(如刚度和韧性等)以利于切片;在聚合过程中,不发生明显的膨胀与收缩;观察样品时,易被电子穿透并能耐受的电子轰击;在高倍放大的图像中不显示本身结构等特征。目前常用的包埋剂是各种环氧树脂。生物样品固定后含有大量水分,而包埋剂多具疏水性质。因此固定的样品在包埋前通常要经过一系列的脱水处理。
(3)切片 超薄切片的制备过程如图2-15所示。切片厚度通常是40~50nm。切片厚度可通过样品杆的金属热膨胀或机械伸缩来控制。切片刀以玻璃或钻石为材料,最常使用的是玻璃刀。切片须捞在覆有支持膜(如 Formvar膜)的载网(铜网或镍网,一般直径3nm)上,用于染色和电镜观察。
(4)染色 电镜样品用重金属盐进行染色。样品中的不同成分对各种重金属盐“染料”有不同的亲和性,如锇酸宜染脂质,柠檬酸铅易染蛋白质,乙酸双氧铀用以染核酸等。当电子束穿过样品时,样品中的金属离子不同程度地散射和吸收电子,在样品的图像上形成明暗差别。因此,电镜下观察到图像只能为黑白图像。
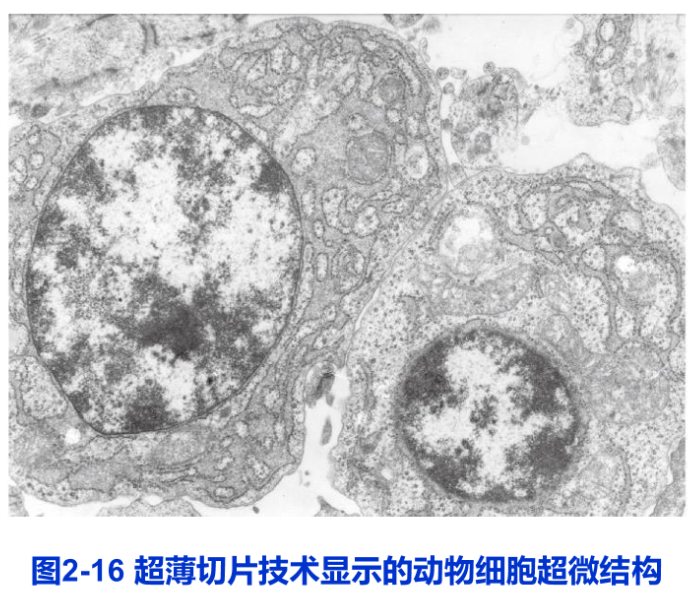
应用超薄切片技术,几乎可以观察各种细胞的超微结构。图2-16即是动物细胞的超薄切片电镜图片。此外,超薄切片技术还可以与细胞化学、免疫化学和原位杂交等技术结合,在超微结构水平上完成蛋白质与核酸等组分的定性、定位和半定量研究。
2.负染色技术
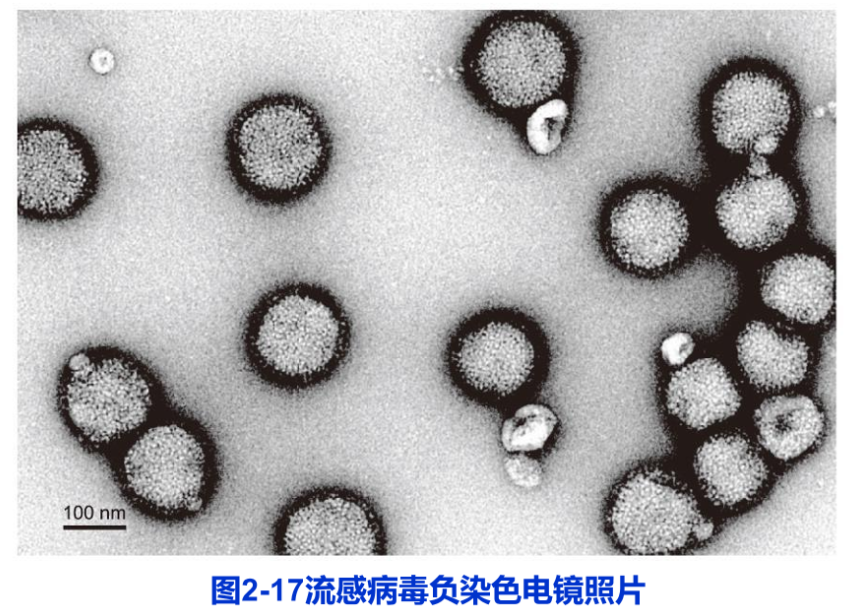
经纯化的细胞组分或结构,如线粒体基粒、核糖体、蛋白颗粒及细胞骨架纤维甚至病毒等可以通过电镜负染色技术(negative staining)显示其精细结构,分辨率可达1.5nm左右。负染色是用重金属盐,如磷钨酸或醋酸双氧铀溶液对铺展在载网上的样品进行染色。吸去多余染料,样品自然干燥后,整个载网上都铺上了一薄层重金属盐,从而衬托出样品的精细结构(图2-17)。
3.冷冻蚀刻技术
冷冻蚀刻技术(freeze etching)的样品制备过程包括冰冻断裂与蚀刻复型两步,因此又称冷冻断裂一蚀刻复型技术。用快速低温冷冻法,在液氮或液氦中将样品迅速冷冻,然后在低温下进行断裂。这时冷冻样品往往从其结构相对“脆弱”的部位(即膜脂双分子层的疏水端)裂开,从而显示出镶嵌在膜脂中的蛋白质颗粒。经过一段时间冰的升华(又称蚀刻),进一步增强图像“浮雕”样的效果。再用铂等重金属进行倾斜喷镀,以形成对应于凹凸断裂面的电子反差。接着,用碳进行垂直喷镀,在断裂面上形成一层连续的碳膜。最后用消化液把生物样品消化掉,将复型膜(碳膜及其被碳膜包裹的,含有图像信息的金属微粒膜)移到载网上,并在电镜下进行观察(图2-18A)。
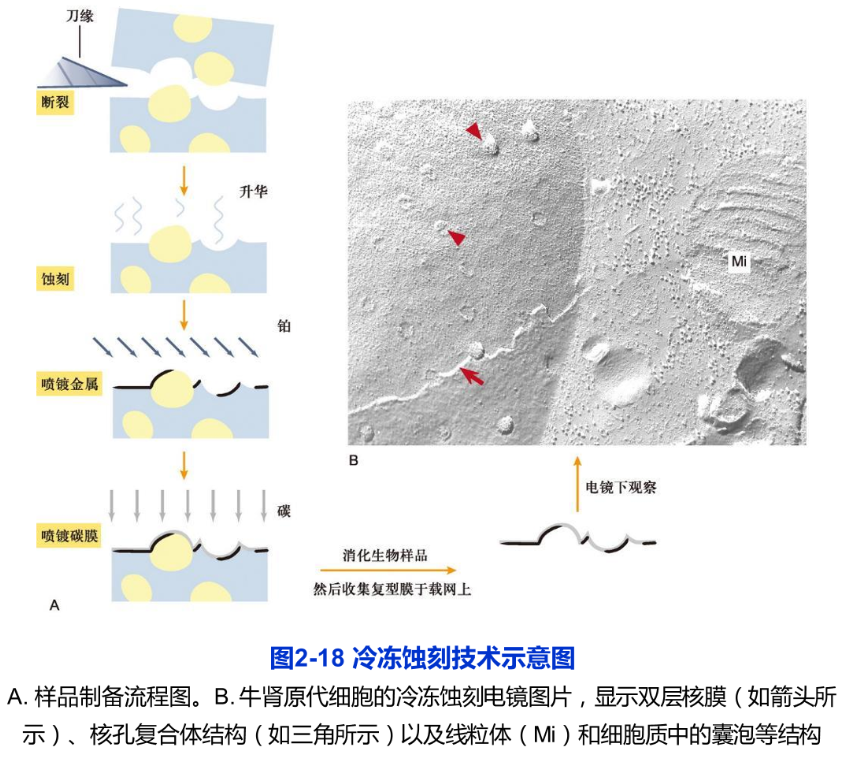
冷冻蚀刻技术主要用来观察膜断裂面上的蛋白质颗粒和膜表面形貌特征,图像富有立体感,样品不需包埋甚至也不需要固定,因而能更好地保持样品的真实结构(图2-18B)。在此基础上又发展了快速冷冻深度蚀刻技术(quick freeze deep etching),该技术主要用于观察胞质中的细胞骨架纤维及其结合蛋白,从而拓宽了这一技术的应用范围。
4.电镜三维重构与低温电镜技术
因为透射电镜利用穿透样品的电子成像,这部分的电子已经携带了样品的内部结构信息,因此我们得到的二维电镜图像中,实际上包含了样品三维结构的全部信息。从二维电镜图像出发,构建出样品的三维图像,称电镜图像的三维重构。从20世纪60年代开始,英国学者A.Kug率先把晶体学的图像数据处理方法用于电镜样品的研究中,成功地获得了负染色样品中T噬菌体的三维结构图像,开创了蛋白质三维结构研究的新方向,因此获得1982年诺贝尔化学奖。
由于染色方法的局限性及负染色后干燥过程对样本身结构细节的损伤,负染色技术制备的生物样品的分辨率只能达到1.5~2nm。经过几十年的不懈努力,人们建立了和发展了低温电镜技术(cryo-electron microscopy),这一技术在细胞内各种生物大分子及其复合物的三维结构分析中,发挥了其他技术不可替代的重要作用,例如膜蛋白(见图3-15“γ-分泌酶复合物三维结构“)、核糖体、30nm染色质(见图9-12“染色质纤维的三维结构”)以及病毒颗粒等。顾名思义,低温电镜技术制备的生物样品,一般不经过固定、包埋、染色和干燥等步骤,而是将样品直接在冷冻剂中快速冷冻,形成100~300nm厚的冰膜,在电镜内-160℃低温下利用相位衬度成像。获得的大量图像还要经过一系列数据处理后,构建出样品的三维结构密度图。
目前主要应用低温电镜单颗粒分析技术(single particle analysis)来研究生物大分子的三维结构,即对包被在冰膜中相同的、分散的、取向随机性的大量颗粒性样品进行拍照和三维重构处理。这是目前最主要的也是发展最快的电镜三维重构方法,其三维结构图像已接近原子尺度的分辨率,为解析生命活动运转中的各种“纳米机器”的结构与作用机理,提供了重要的实验手段。在发展低温电镜技术中贡献巨大的三位科学家J. Dubochet、j. Frank、r. Henderson也因此获得2017年诺贝尔化学奖。
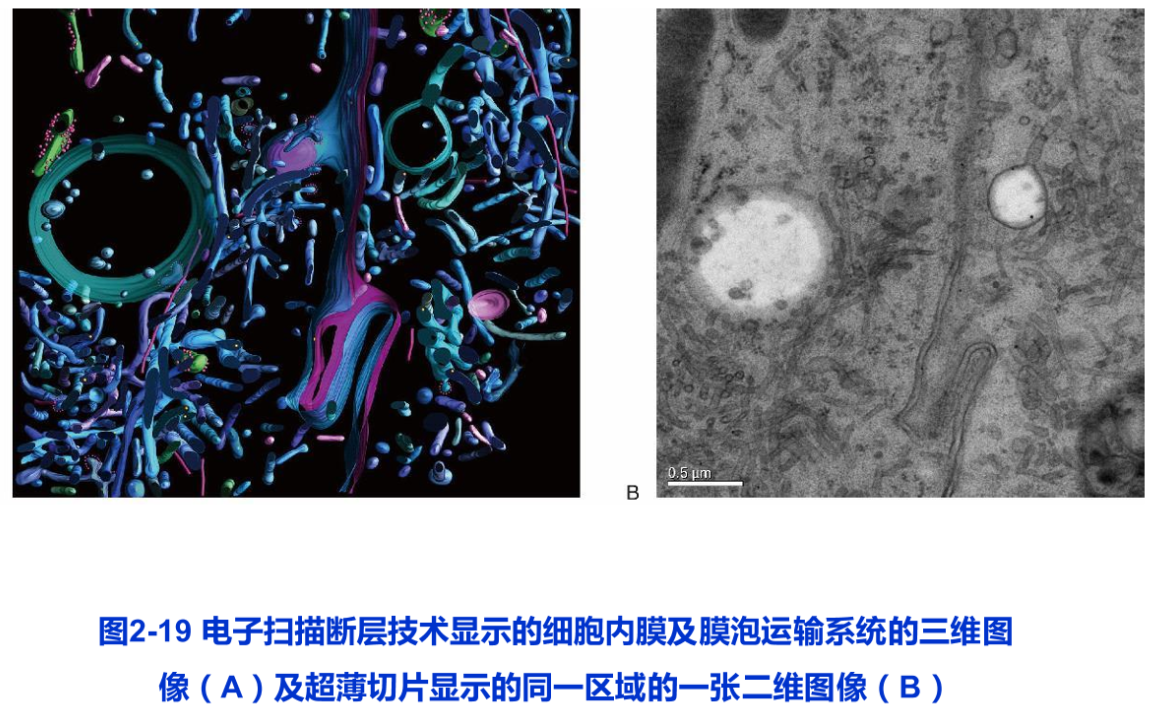
电子断层成像技术(electron tomography)也是利用电镜图像进行三维重构的另一项重要技术。它是对样品中同一物体,在不同的倾角下连续拍照,获得一系列二维投影图片,再重构其三维结构。电子断层成像技术也可用于研究蛋白质复合体的三维结构,但其分辨率明显低于低温电镜单颗粒分析技术。然而这一技术还可用于更大尺度的细胞器、细胞骨架、膜泡运输系统等的三维结构研究,因此在细胞生物学研究中也得到日益广泛的应用。电子断层成像技术可用于冷冻的样品,更多的是用于超薄切片样品(图2-19)。
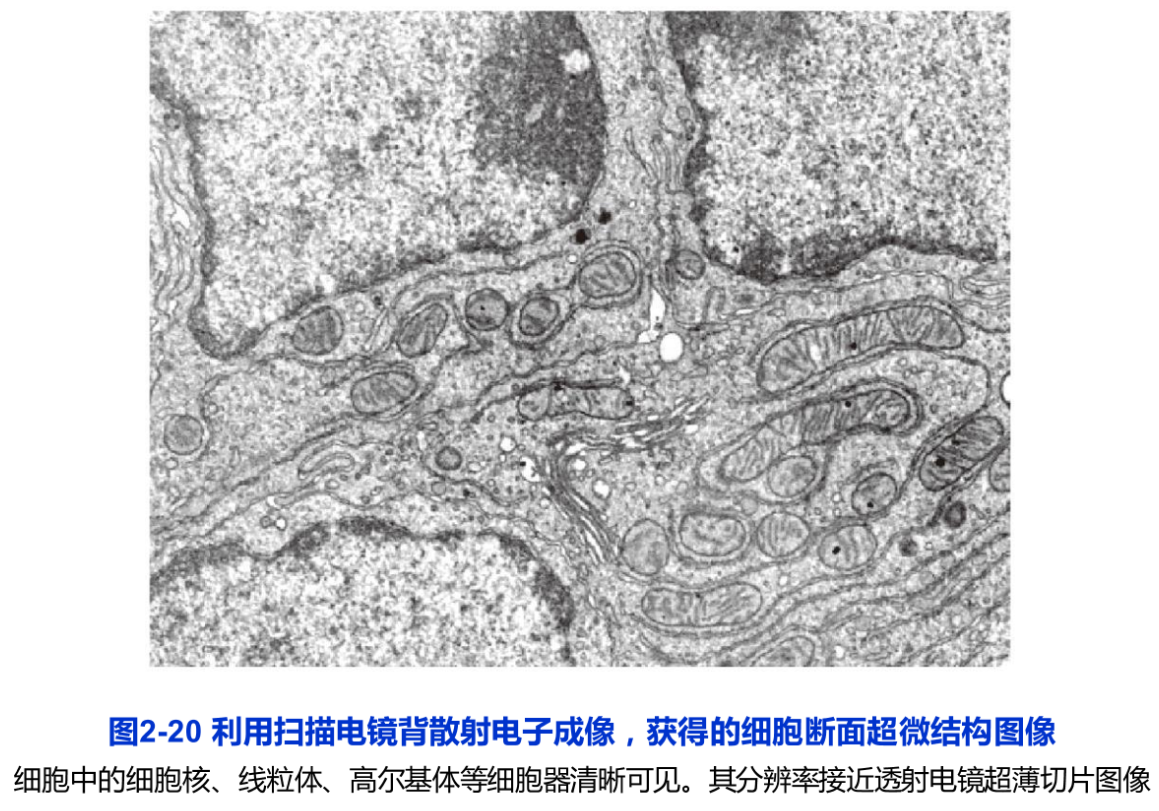
利用扫描电镜背散射电子成像(back scattered electron imaging)技术可以研究细胞、组织等更大尺度的超微结构的三维结构,这在揭示神经元之间的相互关系的脑科学研究中,是一项不可或缺的重要技术。经典的扫描电镜是利用细胞表面的二次电子成像,以观察细胞表面的形貌(见下述“扫描电镜技术”)。而扫描电镜背散射电子图像携带了原子序数的信息,因此,对所观察的组织块染色后,可以显示出如同超薄切片样品的细胞内部结构(图2-20)。在扫描电镜中,做类似超薄切片的处理,同时利用其背散射电子成像,就可以得到一幅幅样品内部的超微结构图像,进而构建成整个组织的三维结构图象。
5.扫描电镜技术
扫描电镜(scanning electron microscope,SEM)问世于20世纪60年代。其电子枪发射出的电子束被磁透镜会聚成极细的电子“探针”,在样品表面进行“扫描”激发样品表面放出二次电子、背散射电子等。二次电子由探测器收集并被闪烁器转变成光信号,其产生的多少与样品表面的形貌相关。这样,通过扫描电镜可以得到样品表面的立体图像信息(图2-21)。
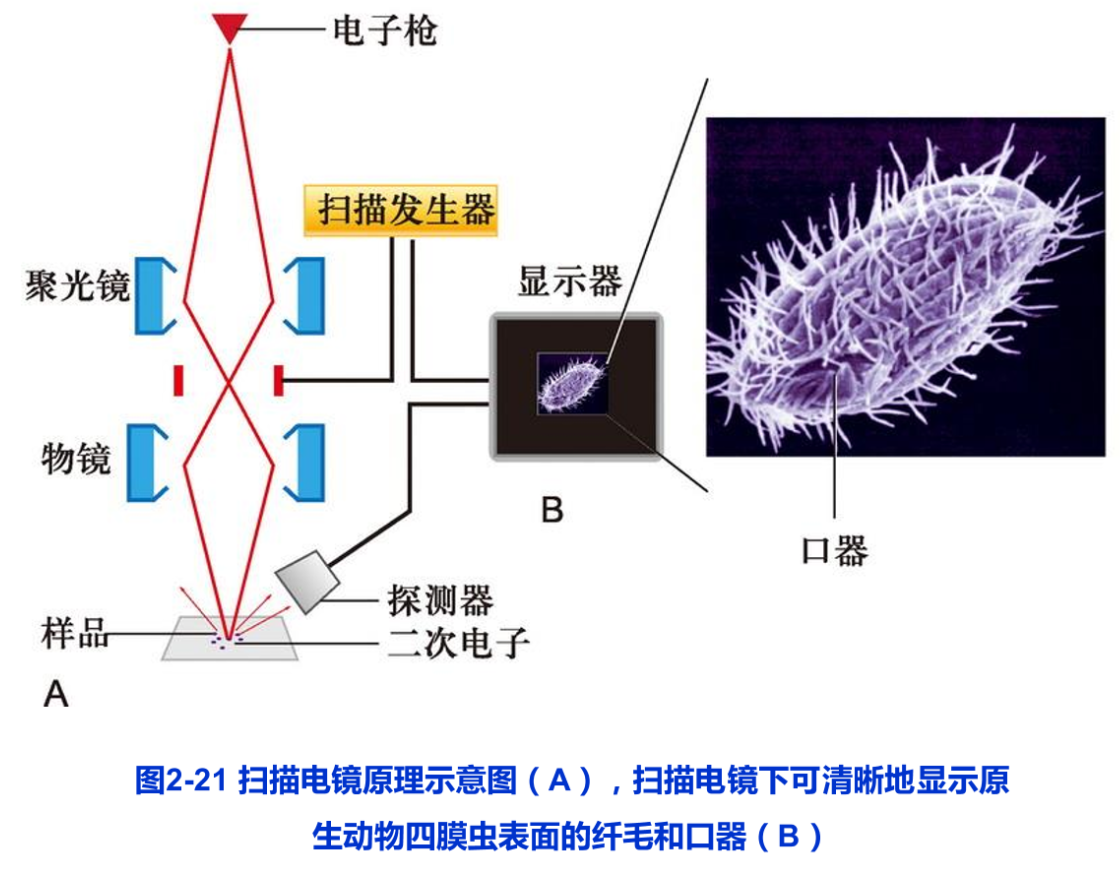
为了保证样品在扫描观察前不发生表面变形,通常需要利用CO2临界点干燥法对样品进行干燥处理。此外,为了得到正确的二次电子信号,样品表面需有良好的导电性。所以样品在观察前还要喷镀一层金膜。
扫描电镜景深长,成像具有强烈的立体感,一般扫描电镜的分辨本领仅为3nm。近些年研制的低压高分辨扫描电镜分辨本领可达0.7nm,可用于观察核孔复合体等更精细的结构。
尽管电子显微镜具有分辨率高这一光学显微镜无可比拟的优越性,但直至目前人们还不能用它来观察活的生物样品,而且难以观察细胞的全貌,因此在很多 研究中仍需要光镜与电镜技术相结合,这就是为什么各种光一电关联技术 (correlative light microscopy and electron microscopy)得到越来越广泛的关注和应用。
三、扫描隧道显微镜
扫描隧道显微镜(scanning tunnel microscope.,STM)是IBM苏黎世实验室的G. Binnig和. Rohrer等人于1981年发明的,它是一种探测微观世界物质表面形貌的仪器。此项发明获得1986年的诺贝尔物理学奖STM的主要原理是利用量子力学中的隧道效应。通常在低电压下,二电极之间具有很大的阻抗,阻止电流通过,称之为势垒;当二电极之间近到一定距离(50nm以内)时,电极之间产生了电流,称隧道电流:这种现象称隧道效应。并且隧道电流(I)和针尖与样品之间的间距(d)呈指数关系[一维近似I∝exp(-2Kd),K为常数],这样d可转化为I的函数而被测定,针尖的位置就可以确定,由此样品的表面形貌也可确定。
STM的主要装置包括实现x、y、z三个方向扫描的压电陶瓷,逼近装置,电子学反馈控制系统和数据采集、处理、显示系统。
STM的主要特点有:①具有原子尺度的高分辨本领,侧分辨率为0.1~0.2nm,纵分辨率可达0.001nm;②可以在真空、大气、液体(接近于生理环境的离子强度)等多种条件下工作,这一点在生物学领域的研究中尤其重要;③非破坏性测量,因为扫描时不接触样品,又没有高能电子束轰击,基本上可避免样品的形变。
目前,STM作为一种新技术,已被广泛应用于生命科学各研究领域。人们已用STM直接观察到DNA、RNA和蛋白质等生物大分子及生物膜、病毒等的结构。与其功能类似的还有原子力显微镜(atomic force microscope,AFM)。原子力显微镜,顾名思义,是利用微小的探针来操纵和测量样品的形貌和力学性质。这个微小的探针在一个悬梁臂弹簧的末端,在与样品直接作用时,悬梁臂的微小位移通过“光杠杆”效应得以放大上千倍投射到光电探测器上,并通过一个反馈通路来控制探针和样品之间的相互作用。原子力显微镜不仅可以精确地测量出样品的形貌,而且还可以测量样品的微观力学性质。其作用力范围在10pN(皮牛)到几十nN(纳牛)之间,恰恰适合于细胞信号分子与配体结合以及蛋白质分子的折叠和展开等研究。
可以预料,STM和AFM等将在纳米生物学乃至纳米科学各领域研究中将发挥越来越重要的作用。