第2讲 先天性免疫系统
HEADS UP!
The innate immune system is a "hard-wired" defense that has evolved over millions of years to recognize pathogens that commonly infect humans. The innate system team includes the complement system of proteins, the professional phagocytes, and natural killer cells. Before they can fight, these warriors must be activated. Cooperation between innate system players is critical to insure a fast and effective response against "everyday" invaders.
INTRODUCTION
For years, immunologists didn't pay much attention to the innate system – because the adaptive system seemed more interesting. However, studies of the adaptive immune system have led to a new appreciation of the role that the innate system plays, not only as a lightning-fast second line of defense (if we count physical barriers as our first defense), but also as an activator and a controller of the adaptive immune system.
It's easy to understand the importance of the innate system's quick response to common invaders if you think about what could happen in an uncontrolled bacterial infection. Imagine that the splinter from your hot tub deck introduced just one bacterium into your tissues. As you know, bacteria multiply very quickly. In fact, a single bacterium doubling in number every 30 minutes could give rise to roughly 100 trillion bacteria in one day. If you've ever worked with bacterial cultures, you know that a 1-liter culture containing one trillion bacteria is so dense you can't see through it. So, a single bacterium proliferating for one day could yield a dense culture of about 100 liters. Now remember that your total blood volume is only about 5 liters, and you can appreciate what an unchecked bacterial infection could do to a human! Without the quick-acting innate immune system to defend us, we would clearly be in big trouble.
THE COMPLEMENT SYSTEM
The complement system is composed of about 20 different proteins that work together to destroy invaders and to signal other immune system players that the attack is on. The complement system is very old. Even sea urchins, which evolved about 700 million years ago, have a complement system. In humans, complement proteins start being made during the first trimester of fetal development, so this important system is ready to go well before a child is born. Indeed, those rare humans born with a defect in one of the major complement proteins usually do not live long before succumbing to infection.
When I first read about the complement system, I thought it was way too complicated to even bother understanding. But as I studied it further, I began to realize that it is really quite simple and elegant. As with just about everything else in the immune defense, the complement system must be activated before it can function, and there are three ways this can happen. The first, the so-called "classical" pathway, depends on antibodies for activation, so we'll save that for a later lecture. Because the way the complement system functions is independent of how it is activated, you won't miss much by waiting to hear about the antibody-dependent path-way of activation.
The alternative pathway
The second way the complement system can be activated is called the alternative pathway – although the alternative pathway certainly evolved before the classical pathway. Immunologists call antibody-dependent activation "classical," simply because it happened to have been discovered first.
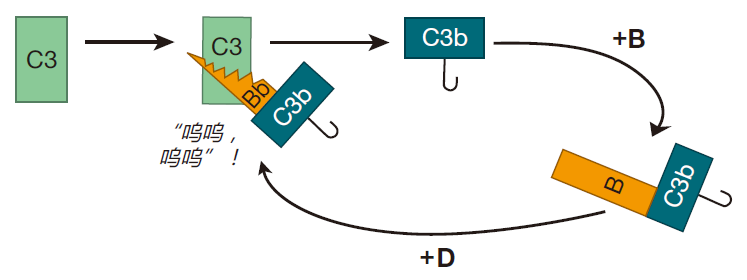
The proteins that make up the complement system are produced mainly by the liver, and are present at high concentrations in blood and tissues. The most abundant complement protein is called C3, and in the human body, C3 molecules are continually being broken into two smaller proteins. One of the protein fragments created by this "spontaneous" cleavage, C3b, is very reactive, and can bind to either of two common chemical groups (amino or hydroxyl groups). Because many of the proteins and carbohydrates that make up the surfaces of invaders (e.g., bacterial cells) have amino or hydroxyl groups, there are lots of targets for these little C3b "grenades."
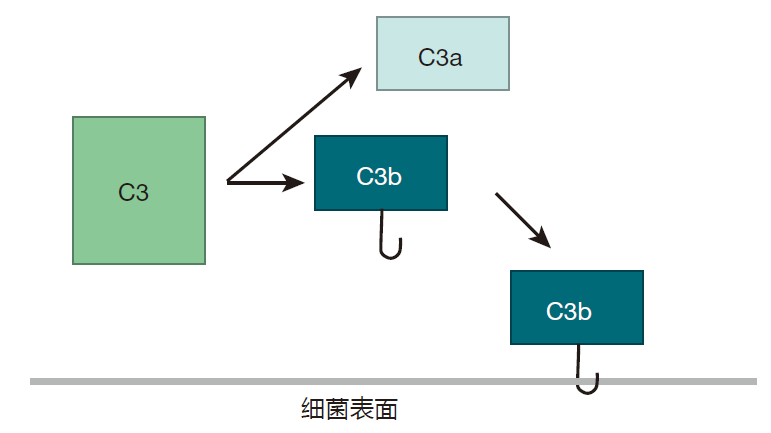
If C3b doesn't find one of these chemical groups to react with within about 60 microseconds, it is neutralized by binding to a water molecule, and the game is over. This means that the spontaneously clipped C3 molecule has to be right up close to the surface of the invading cell in order for the complement cascade to continue. Once C3b is stabilized by reacting with a molecule on the cell surface, another complement protein, B, binds to C3b. Then complement protein D comes along and clips off part of B to yield C3bBb.
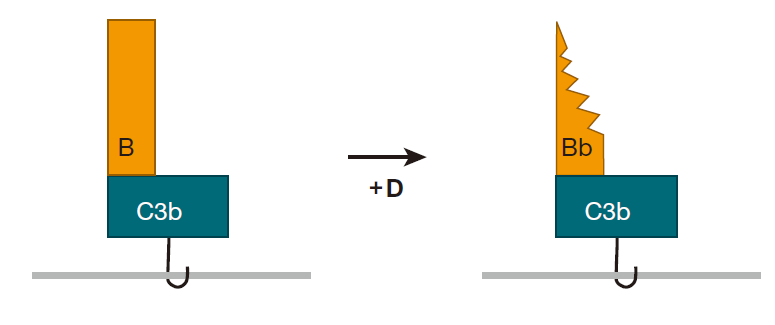
Once a bacterium has this C3bBb molecule glued to its surface, the fun really begins, because C3bBb acts like a "chain saw" that can cut other C3 proteins and convert them to C3b. Consequently, C3 molecules that are in the neighborhood don't have to wait for spontaneous clipping events to convert them to C3b – the C3bBb molecule (called a convertase) can do the job very efficiently. And once another C3 molecule has been clipped, it too can bind to an amino or hydroxyl group on the surface of the bacterium.
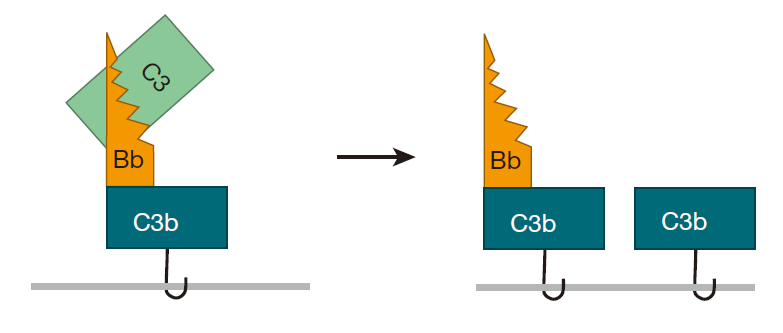
This process can continue, and pretty soon there are lots of C3b molecules attached to the surface of the target bacterium –, each of which can form a C3bBb convertase – which can then cut even more C3 molecules. All this attaching and cutting sets up a positive feedback loop, and the whole process just snowballs.
Once C3b is bound to the surface of a bacterium, the complement cascade can proceed further. The C3bBb chain saw can bind to another molecule of C3b, and together they can clip a complement protein, C5, into two pieces. One of these pieces, C5b, can then combine with other complement proteins (C6, C7, C8, and C9) to make a membrane attack complex (MAC). To form this structure, C5b, C6, C7, and C8 form a "stalk" that anchors the complex in the bacterial cell membrane. Then C9 proteins are added to make a channel that opens up a hole in the surface of the bacterium. And once a bacterium has a hole in its surface, it's toast!
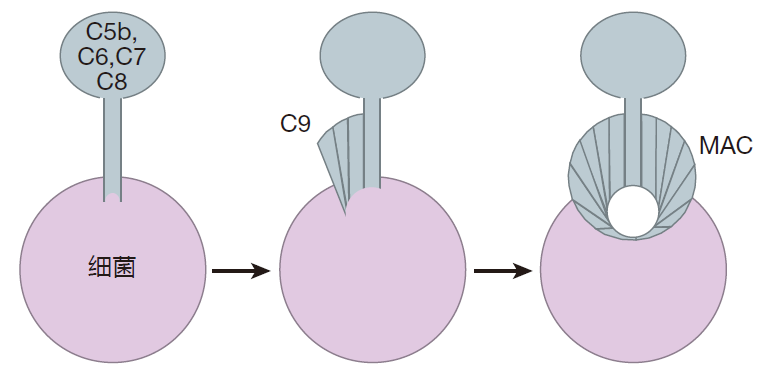
I have used a bacterium as our "model pathogen," but the complement system also can defend against other invaders such as parasites and even some viruses. For example, complement proteins can poke holes in enveloped viruses (e.g., the AIDS virus) by constructing membrane attack complexes on the surface of the virus.
Now, you may be thinking: With these grenades going off all over the place, why doesn't the complement system form membrane attack complexes on the surface of our own cells? The answer is that human cells are equipped with many safeguards to keep this from happening. In fact, there are about as many proteins devoted to controlling the complement system as there are proteins in the system itself! For instance, the complement fragment, C3b, can be clipped to an inactive form by proteins in the blood, and this clipping is accelerated by an enzyme (MCP) that is present on the surface of human cells. There is also a protein on human cells called decay accelerating factor (DAF), which accelerates the destruction of the convertase, C3bBb, by other blood proteins. This can keep the positive feedback loop from getting started. And yet another cell-surface protein, CD59 (also called protectin), prevents the incorporation of C9 molecules into nascent MACs.
An interesting story illustrates why these safeguards are so important. Transplant surgeons don't have enough human organs to satisfy the demand for transplantation, so they are considering using organs from animals. One of the hot candidates for an organ donor is the pig, because pigs are cheap to raise and some of their organs are about the same size as those of humans. As a warm-up for human transplantation, surgeons decided to transplant a pig organ into a baboon. This experiment was not a big success! Almost immediately, the baboon's immune system began to attack the organ, and within minutes the transplanted organ was a bloody pulp. The culprit? The complement system. It turns out that the pig versions of DAF and CD59 don't work to control primate complement, so the unprotected pig organ was vulnerable to attack by the baboon's complement system.
This story highlights two important features of the complement system. First, the complement system works very fast. Complement proteins are present at high concentrations in blood and in tissues, and they are ready to go against any invader that has a surface with a spare hydroxyl or amino group. A second characteristic of this system is that if a cell surface is not protected, it will be attacked by complement. In fact, the picture you should have is that the complement system is continually drop-ping these little grenades, and any unprotected surface will be a target. In this system, the default option is death!
The lectin activation pathway
In addition to the classical (antibody-dependent) and alternative (antibody-independent) pathways of complement activation, there is a third pathway that may be the most important activation pathway of all: the lectin activation pathway. The central player in this pathway is a protein that is produced mainly in the liver, and which is present in moderate concentrations in the blood and tissues. This protein is called mannose-binding lectin (MBL). A lectin is a protein that is able to bind to a carbohydrate molecule, and mannose is a carbohydrate molecule found on the surface of many common pathogens. For example, MBL has been shown to bind to yeasts such as Candida albicans; to viruses such as HIV-1 and influenza A; to many bacteria, including Salmonella and Streptococcus; and to parasites such as Leishmania. In contrast, mannose-binding lectin does not bind to the carbohydrates found on healthy human cells and tissues. This is an example of an important strategy employed by the innate system: The innate system mainly focuses on patterns of carbohydrates and fats that are found on the surface of common pathogens, but not on the surface of human cells.
The way mannose-binding lectin works to activate the complement system is very simple. In the blood, MBL binds to another protein called MASP. Then, when the mannose-binding lectin grabs its target (mannose on the surface of a bacterium, for example), the MASP protein functions like a convertase to clip C3 complement proteins to make C3b. Because C3 is so abundant in the blood, this happens very efficiently. The C3b fragments can then bind to the surface of the bacterium, and the complement chain reaction we just discussed will be off and running. So, whereas the alternative activation pathway is spontaneous, and can be visualized as complement grenades going off randomly here and there to destroy any unprotected surface, lectin activation can be thought of as complement "smart bombs" that are targeted by mannose-binding lectins.
Other complement system functions
In addition to building membrane attack complexes, the complement system has two other important functions. When C3b has attached itself to the surface of an invader, it can be clipped by a serum protein to produce a smaller fragment, iC3b. The "i" prefix denotes that this cleaved protein is now inactive for making MACs. However, it is still glued to the invader, and it can prepare the invader for phagocytosis (i.e., can opsonize it) in much the same way that invaders can be opsonized by antibodies. On the surface of phagocytes (e.g., macrophages) are receptors that can bind to iC3b, and the binding of iC3b-opsonized invaders facilitates phagocytosis. Many invaders have surfaces that are rather "slimy," making them difficult for macrophages to grasp. However, when these slippery invaders are coated with complement fragments, phagocytes can get a better grip. Thus, a second function of complement is to decorate the surfaces of invaders, thereby acting like a "poor man's antibody" in opsonization.
The complement system has a third important function: Fragments of complement proteins can serve as chemoattractants – chemicals that recruit other immune system players to the battle site. For example, C3a and C5a are the pieces of C3 and C5 that are clipped off when C3b and C5b are made (let nothing be wasted!). These fragments don't bind to the surface of invaders. Rather, they are set free in the tissues where they function as chemoattractants. C5a is an especially powerful chemoattractant for macrophages, and can activate them so that they become more potent killers. Interestingly, these fragments, C3a and C5a, are called anaphylatoxins, because they can contribute to anaphylactic shock – something we will talk about in another lecture.
So the complement system is quite multifunctional: It can destroy invaders by building membrane attack complexes; it can tag intruders for destruction by phagocytes; it can alert other cells that we are being attacked and direct them to the battle scene; and it can help activate them. Most importantly, it can do all these things very fast.
THE PROFESSIONAL PHAGOCYTES
Professional phagocytes comprise the second arm of the innate system. These cells are called "professional" because they make their living mainly by eating (phagocytosis). The most important professional phagocytes are the macrophages and the neutrophils.
Macrophages – immune system sentinels
Macrophages are found under your skin, where they provide protection against invaders which penetrate this barrier and gain entry into your tissues (e.g., as the result of a wound or a burn). Macrophages also are present in your lungs, where they defend against inhaled microbes. Still other macrophages reside in the tissues that surround your intestines. There they lie in wait for microbial invaders you have ingested, which have escaped the confines of your intestines and have entered your tissues. Indeed, macrophages are sentinel cells which can be found just below the surface in all areas of your body exposed to the outside world – areas that are prime targets for microbial infection. Macrophages are present in most tissues before birth, so are already "on duty" when a baby is born. Later, in response to an infection, these tissue-resident macrophages can proliferate, and monocytes also can be recruited from the bone marrow, enter infected tissues, and mature into macrophages.
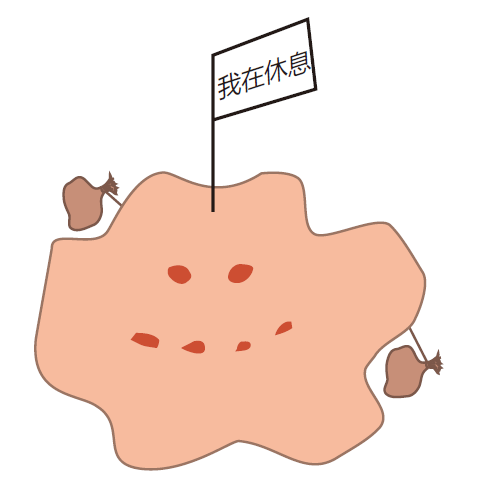
Macrophages can exist in three stages of readiness. In tissues, they usually are found just lounging and slowly proliferating. In this "resting" state, they function primarily as garbage collectors, taking sips of whatever is around them, and keeping our tissues free of debris. About one million cells die per second in an adult human, so macrophages have a lot of tidying up to do. Dying cells give off "find me" signals that attract macrophages, bringing them close enough to recognize "eat me" signals displayed on the surface of cells when they die. Healthy cells, on the other hand, display "don't eat me" signals on their surface to protect them from macrophage ingestion.
While resting, macrophages express very few class II MHC molecules on their surface, so they aren't much good at presenting antigen to helper T cells. This makes sense. Why would they want to present garbage anyway? For the average macrophage, life is pretty boring. They live for months in tissues and just collect garbage.
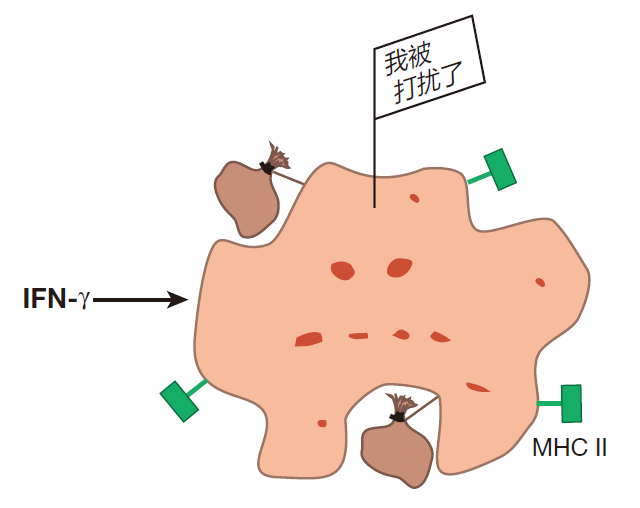
Every once in a while, however, some of these resting macrophages receive signals which alert them that the barrier defense has been penetrated, and that there are intruders in the area. When this happens, they become activated (or "primed," as immunologists usually say). In this state, macrophages take larger gulps and upregulate expression of class II MHC molecules. Now a macrophage can function as an antigen presenting cell, and when it engulfs invaders, it can use its class II MHC molecules to display fragments of the invaders' proteins for helper T cells to see. Although a number of different signals can prime a resting macrophage, the best studied is an intercellular communication molecule (cytokine) called interferon gamma (IFN-γ). This cytokine is produced mainly by helper T cells and natural killer cells.
In the primed state, macrophages are good antigen presenters and reasonably good killers. However, there is an even higher state of readiness, "hyperactivation," which they can attain if they receive a direct signal from an invader. Such a signal can be conveyed, for example, by a molecule called lipopolysaccharide (LPS). LPS, a component of the outer cell membrane of Gram-negative bacteria such as Escherichia coli, can be shed by these bacteria, and can bind to receptors on the surface of primed macrophages. Macrophages also have receptors for mannose. When receptors on the surface of the macrophage bind to "danger signals" such as LPS or mannose, the macrophage knows for sure that there has been an invasion. Faced with this realization, the macrophage stops proliferating, and focuses its attention on killing. In the hyperactive state, macrophages grow larger and increase their rate of phagocytosis. In fact, they become so large and phagocytic that they can ingest invaders as big as unicellular parasites. Hyperactivated macrophages also produce and secrete another cytokine, tumor necrosis factor (TNF). This cytokine can kill tumor cells and virus-infected cells, and can help activate other immune system warriors.
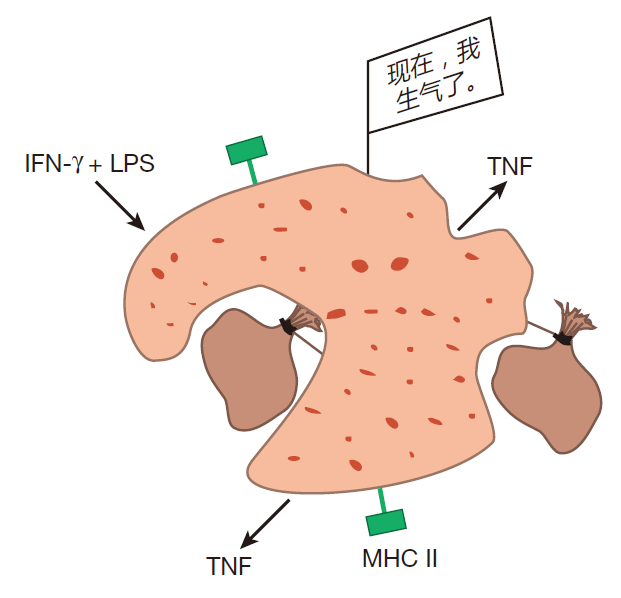
Inside a hyperactivated macrophage the number of lysosomes increases so that the destruction of ingested invaders becomes more efficient. In addition, hyperactivated macrophages increase production of reactive oxygen molecules such as hydrogen peroxide. You know what peroxide can do to hair, so you can imagine what it might do to a bacterium! Finally, when hyperactivated, a macrophage can dump the contents of its lysosomes onto multicellular parasites, enabling it to destroy invaders that are too large to "eat." Yes, a hyperactivated macrophage is a killing machine!
So a macrophage is a versatile cell. It can function as a garbage collector, as an antigen presenting cell, or as a vicious killer – depending on its activation level. However, you shouldn't get the impression that macrophages have three "gears." Nothing in immunology has gears, and the activation state of a macrophage is a continuum that really depends on the type and the strength of the activation signals it receives.
Usually, macrophages are able to deal with small attacks. However, when invaders are numerous, macrophages risk being overpowered, and in those cases, macrophages call for backup. The most common reinforcement for battling macrophages is a cell called a neutrophil. Indeed, although the macrophage is unmatched in versatility, the most important of the professional phagocytes is probably the neutrophil.
Neutrophils – the immune system's foot soldiers
All of our cells receive their nutrients from the blood, and consequently no cell is more than about the thickness of a fingernail from a blood vessel. If a cell is farther away than that, it will die of starvation. Because our tissues are laced with blood vessels, blood is the perfect vehicle for bringing reinforcements to parts of the body that are under attack. And circulating through our veins and arteries are about 20 billion neutrophils. In contrast to macrophages, which can be thought of as sentinels, neutrophils are more like "foot soldiers." Their job is to "kill things and break stuff," and they are really good at this.
Neutrophils live a very short time. In fact, they come out of the bone marrow programmed to die in an average of about five days. In contrast to macrophages, neutrophils are not antigen presenting cells. They are professional killers which are "on call" from the blood.
Once they have been summoned, it only takes neutrophils about half an hour to exit the blood and become fully activated. In this state, neutrophils are incredibly phagocytic, and once their prey has been taken inside, a whole battery of powerful chemicals awaits the unlucky "guest." Neutrophils also produce battle cytokines (e.g., TNF) that can alert other immune system cells. And most importantly, activated neutrophils give off destructive chemicals which are pre-made and stored inside the neutrophil until needed. These chemicals can turn tissues into a "toxic soup" that is lethal to invading microbes. Indeed, neutrophils are unique in that they are the only immune system cells that are "licensed" to liquify both cells and connective tissue.
My friend Dan Tenen studies neutrophils. Another friend, Linda Clayton, who experiments with T cells, likes to kid him by asking, "Why do you bother studying neutrophils, Dan? All they do is dive into pus and die!" She's right, of course. Pus is mainly dead neutrophils. However, Dan reminds her that humans can live for long periods without her fancy T cells, but without his neutrophils they will succumb to infection and die within a matter of days.
Now, why do you think things are set up so that macrophages are very long lived, yet neutrophils live only a few days? Doesn't that seem wasteful? Why not let neutrophils enjoy a long life, just like macrophages? That's right! It would be too dangerous. Neutrophils come out of blood vessels ready to kill, and in the course of this killing, there is always damage to normal tissues. So to limit collateral damage, neutrophils are programmed to be short-lived. If the battle requires additional neutrophils, more can be recruited from the blood – there are plenty of them there. Indeed, neutrophils represent about 70% of the circulating white blood cells. In contrast, because macrophages act as sentinels that watch for invaders and signal an attack, it makes sense that macrophages should have a long life out in the tissues.
It has been known for a long time that neutrophils are voracious phagocytes, and that they can give off chemicals that destroy both invaders and tissues. Recently, however, it was discovered that under certain conditions, some dying neutrophils can release web-like structures called neutrophil extracellular traps (NETs). These NETs are composed of cellular DNA that is coated with proteins derived from the granules in which neutrophils store the chemicals they use to do their destructive work. In the laboratory, NETs can trap or kill bacteria, viruses, fungi, and parasites. Nevertheless, it is not yet clear what triggers neutrophils to release these NETs, or how important NETs are for the immune defense. Although it is possible that NETs play a role in protecting us against certain invaders, neutrophil function usually is tightly controlled to limit unwanted tissue damage. Consequently, it seems counterintuitive that the inflammation and tissue damage caused by NETs would be a good thing. Indeed, much of the research on neutrophil extracellular traps centers on discovering the possible role this phenomenon might have in disease.
How neutrophils exit the blood
You may be wondering: If neutrophils are all that dangerous, how do they know when to leave the blood stream and where to go? It certainly wouldn't do to have neutrophils leave the blood and become activated just any old place. No indeed, and the way this works is very clever. Inside blood vessels, neutrophils exist in an inactive state, and they are swept along by the blood at high speed – about 1,000 microns per second. If you're the size of a neutrophil, that's really fast.
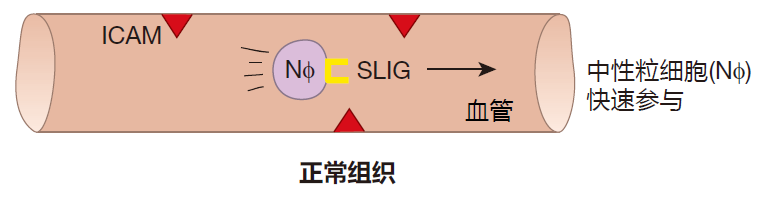
In this sketch, you will notice that there is a protein, intercellular adhesion molecule (ICAM), which is expressed on the surface of the endothelial cells that line blood vessels. There is also another adhesion molecule called selectin ligand (SLIG) that is expressed on the surface of neutrophils. As you can see, however, these two adhesion molecules are not "partners," so they don't bind to each other, and the neutrophil is free to zip along with the flowing blood.
Now imagine that you get a splinter in your big toe, and that the bacteria on the splinter activate macrophages which are standing guard in the tissues of your foot. These activated macrophages give off cytokines, inter-leukin 1 (IL-1) and TNF, which signal that an invasion has begun. When endothelial cells that line nearby blood vessels receive these alarm signals, they begin to express a new protein on their surface called selectin (SEL). It normally takes about six hours for this protein to be made and transported to the surface of endothelial cells. Selectin is the adhesion partner for selectin ligand, so when selectin is expressed on the endothelial cell surface, it functions like Velcro to grab neutrophils as they fly by. However, this interaction between selectin and its ligand is only strong enough to cause neutrophils to slow down and roll along the inner surface of the blood vessel.
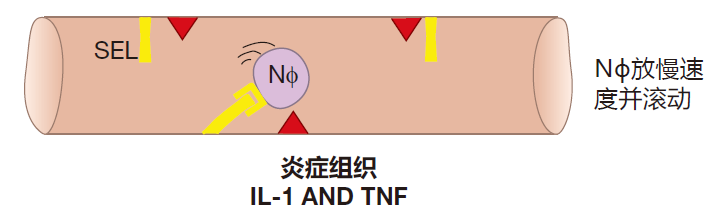
As a neutrophil rolls, it "sniffs." What it's sniffing for is a signal that there is a battle (an inflammatory reaction) going on in the tissues. The complement fragment C5a and LPS are two of the inflammatory signals that a neutrophil recognizes. When it receives such signals, the neutrophil rushes a new protein called integrin (INT) to its surface. To make rapid surface expression of integrin possible, a lot of this protein is made in advance by the neutrophil and stored inside the cell until needed. This quick reaction is important, because the neutrophil hasn't stopped – it's still rolling along. If it rolls too far, it will leave the region where selectin is expressed, and will then start to zoom along again at "blood speed."
When integrin appears on the neutrophil's surface, it interacts with its binding partner, ICAM, which always is expressed on the surface of endothelial cells. This interaction is very strong and causes the neutrophil to stop rolling.
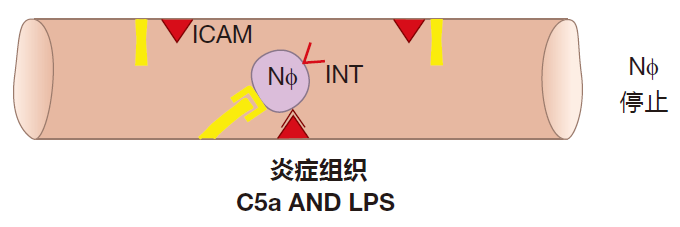
Once a neutrophil has stopped, it can be encouraged by chemoattractants to pry apart the endothelial cells that line the blood vessels, exit into the tissues, and migrate to the site of inflammation. These chemoattractants include the complement fragment C5a and fragments of bacterial proteins called f-met peptides. Bacterial proteins begin with a special initiator amino acid called formyl methionine (f-met). In human cells, only mitochondria produce proteins with this initiator, so less than 0.1% of all human proteins contain this amino acid. Bacteria secrete f-met peptides, and macrophages burp up these protein fragments when they ingest bacteria. Consequently, C5a and f-met peptides function as "find me" signals to help phagocytes locate invaders which have been identified as dangerous by the innate system. And as they travel through the tissues, neutrophils can be activated by cytokines such as TNF. As a result, they arrive at the battle scene ready to kill.
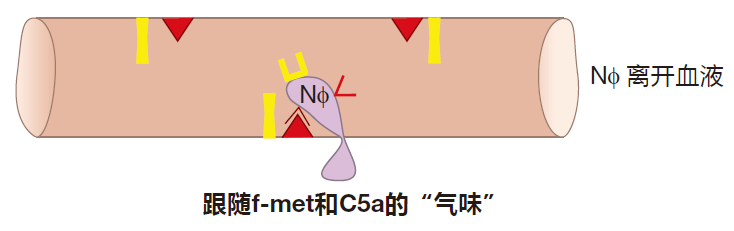
Neutrophil logic
This system – which involves selectin–selectin ligand binding to make the neutrophil roll, integrin–ICAM interactions to stop the neutrophil, and chemoattractants and their receptors to encourage the neutrophil to exit from the blood – may seem overly complicated. Wouldn't it be simpler just to have one pair of adhesion molecules (say, selectin and its ligand) do all three things? Yes, it would be simpler, but it would also be very dangerous. In a human there are about 100 billion endothelial cells. Suppose one of them gets a little crazy and begins to express a lot of selectin on its surface. If selectin binding were the only requirement, neutrophils could empty out of the blood into normal tissues where they could do terrible damage. Having three types of molecules which must be expressed before neutrophils can exit the blood and spring into action helps make the system fail-safe.
You remember I mentioned that to completely upregulate expression of that first cellular adhesion molecule, selectin, takes about six hours. Doesn't this seem a bit too leisurely? Wouldn't it be better to begin recruiting neutrophils from the blood just as soon as a macrophage senses danger? Not really. Before you start to recruit reinforcements, you want to be sure that the attack is serious. If a macrophage encounters only a few invaders, it can usually handle the situation without help in a short time. Summoning neutrophils would only lead to unnecessary tissue damage. In contrast, a major invasion involving many macrophages can go on for days. The sustained expression of alarm cytokines from many macrophages engaged in battle is required to upregulate selectin expression, and this insures that more troops will be summoned only when they really are needed.
Neutrophils are not the only blood cells that need to exit the blood and enter tissues. For example, mast cells, which are involved in protection against parasites, must exit the blood at the site of a parasitic infection. Monocytes, which can mature into tissue macrophages, need to leave the blood stream at appropriate places. And activated B cells and T cells must be dispatched to sites of infection. This whole business is like a postal system in which there are trillions of packages (immune system cells) that must be delivered to the correct destinations. This delivery problem is solved by using the same basic strategy that works so well for neutrophils. The key feature of the immune system's "postal service" is that the Velcro-like molecules which cause the cells to roll and stop are different from cell type to cell type and destination to destination. As a result, these cellular adhesion molecules actually serve as "zip codes" to insure that cells are delivered to the appropriate locations. Indeed, the selectins and their ligands are really families of molecules, and only certain members of the selectin family will pair up with certain members of the selectin ligand family. The same is true of the integrins and their ligands. Because of this two-digit zip code (type of selectin, type of integrin), there are enough "addresses" available to send the many different immune system cells to all the right places. Because immune system cells are equipped with particular adhesion molecules, and their intended destinations express the corresponding adhesion partners, the different types of immune system cells will roll, stop, and exit the blood exactly where they are needed.
HOW IMMUNE SYSTEM SENTINELS RECOGNIZE INVADERS
Before immune system cells such as macrophages can spring into action, they must first recognize that there has been an invasion. But how do they do this? The answer is that immune system cells come equipped with an array of pattern-recognition receptors (PRRs) which are designed to recognize "danger signals" associated with a microbial attack. Altogether, there are more than 20 different PRRs that are expressed by various types of immune system cells. When their pattern-recognition receptors detect invaders, warrior cells such as macrophages are activated, and battle cytokines are produced which alert and activate other immune system cells.
Some pattern-recognition receptors detect pathogen-associated molecular patterns (PAMPs) that are characteristic of broad classes of invaders. For example, the LPS molecule of Gram-negative bacteria is an important PAMP. Other PRRs are tuned to recognize damage-associated molecular patterns (DAMPs). Molecules that function as DAMPs are normally intracellular, but are released by dying cells (e.g., cells killed by viruses).
Consequently, DAMPs can alert the immune system to widespread cellular death associated with an infection. DAMPs are important because they allow immune system cells to respond to pathogens for which there is no specific PRR, including new pathogens that have not been encountered before.
The PRRs about which most is known are the Toll-like receptors (TLRs). So far, ten human TLRs have been discovered, and different cells express different combinations of these TLRs. Some TLRs are displayed on the cell surface, where they respond to invaders that are outside the cell. For example, TLR4 is used by macrophages to sense the presence of LPS. TLR4 is anchored in the macrophage's plasma membrane, and points outward to sense bacterial invaders in the external environment.
Other PRRs are found inside cells, and these receptors detect invaders that are within these cells. For instance, when invaders are phagocytosed, they end up in phago-lysosomes, where they are eventually destroyed. During this destruction, their "coats" are stripped off to reveal what's inside them. Some Toll-like receptors (e.g., TLR7 and TLR9) are located in the membranes of phago-lysosomes. These pattern-recognition receptors point inward into the phago-lysosome so that they can alert the cell to the presence of viruses or bacteria that have been phago-cytosed. TLR7 detects the single-stranded RNA of viruses such as influenza and HIV-1, whereas TLR9 recognizes the double-stranded DNA of bacteria and herpes simplex virus.
Pattern-recognition receptors have two important properties. First, PRRs recognize general characteristics of classes of invaders – not just a single invader. For example, LPS is a common component of bacterial cell membranes, and single-stranded RNA is found in many viruses. Consequently, TLR4 can detect invasions by many different types of bacteria (those with LPS in their cell membrane), and TLR7 can alert cells to attacks by many different viruses (those which carry their genetic information in the form of single-stranded RNA). So in contrast to B cell receptors and T cell receptors, which are specific for each invader, pattern-recognition receptors are "economical" in the sense that each one can identify many different pathogens.
There is a second important characteristic of PRRs: The patterns they recognize represent structural features which are so important to the pathogen that they cannot easily be altered by mutation to avoid detection. For instance, the region of the LPS molecule which TLR4 recognizes is indispensable for constructing bacterial outer membranes. Consequently, a bacterium would be in big trouble if that part of the LPS molecule were mutated to try to evade detection by TLR4.
HOW THE INNATE IMMUNE SYSTEM DEALS WITH VIRUSES
When a virus infects a human cell it takes over the cell's machinery and uses it to produce many more copies of the virus. Eventually, the newly made viruses burst out of the infected cell, and go on to infect other cells in the neighborhood. We have already discussed some of the weapons the innate system can use to defend against viruses while they are outside of cells. For example, proteins of the complement system can opsonize viruses for phagocytosis by macrophages and neutrophils, and complement proteins can destroy some viruses. However, once a virus has entered a cell to begin its reproductive cycle, these weapons are ineffective.
The interferon system
Fortunately, there are other innate system weapons which are useful against virus-infected cells. Indeed, the innate system weapon that viruses fear most is the interferon system. This defense is so potent that most viruses have evolved ways to try to fend off the interferon system – at least long enough for the virus to reproduce and spread to a new host.
When their pattern-recognition receptors detect a virus attack, cells can produce "warning proteins" called interferon alpha (IFN-α) and interferon beta (IFN-β) – proteins that can "interfere" with viral reproduction. IFN-α and IFN-β are called type I interferons to distinguish them from IFN-γ, mentioned earlier, which is a type II interferon. Most human cells quickly produce type I interferons when they are attacked by a virus and have receptors on their surface for these interferon proteins. Consequently, the interferon produced by these cells can actually bind to receptors on the infected cell itself. This binding can result in the production of several hundred antiviral proteins that act to reduce the amount of virus produced by the infection. Even better, when the type I interferons produced by virus-infected cells bind to interferon receptors on nearby cells, those cells are warned that there are viruses in the area, and that they may soon be attacked. As a result of this early warning, the alerted cells turn on expression of antiviral genes and prepare to commit suicide if the virus does infect them.
The elegant part of the interferon warning system is that although the binding of interferon to its receptors prepares an uninfected cell for a viral attack, that cell continues to do business as usual unless an attack actually occurs. The alerted cell will only commit suicide if it is infected by a virus. Of course, for the cell, this is an altruistic act – because both the cell and the virus die together. Nevertheless, this "beneficial suicide" does prevent the virus from reproducing and infecting other cells. And if the attack does not come, the warned cell eventually "stands down" from its state of readiness.
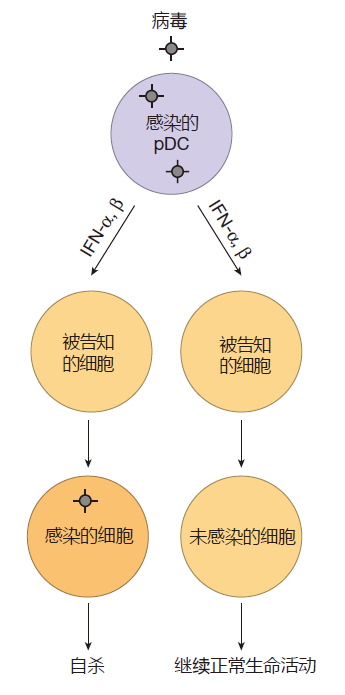
Although many cell types can produce type I interferons, the "King of Interferon" by far is a white blood cell called a plasmacytoid dendritic cell (pDC). Human pDCs use TLR7 and TLR9 to detect viral RNA and DNA, respectively, and when either of these PRRs is engaged, pDCs dedicate more than half of their protein-making capacity to making interferon. As a result, a plasmacytoid dendritic cell can make up to 1,000 times as much type I interferonper day as any other cell type! Consequently, these "interferon factories" can play a critical role in the innate system's defense against viruses, especially early in a viral infection.
Natural killer cells
There is another important player on the innate immune system team which can help defend against a viral infection: the natural killer (NK) cell. Indeed, humans with genetic defects which make them deficient in NK cell function have great difficulty controlling herpes virus and human papillomavirus infections.
Natural killer cells mature in the bone marrow and, when they are not responding to an infection, are short-lived, with a half life of only about a week. Most NK cells are found in the blood or in the spleen and liver (two organs that store blood), and relatively few NK cells reside in tissues that are not under attack. So, like neutrophils, natural killer cells are mostly on call. NK cells use the "roll, stop, exit" strategy to leave the blood and enter tissues at sites of infection – and once in the tissues, they proliferate rapidly to build up their numbers.
When they reach the battleground, natural killer cells can play two roles in defending us against infections. First, NK cells can give off cytokines such as IFN-γ that help with the defense. Like macrophages, NK cells can exist in several stages of readiness. Resting NK cells produce some cytokines and can kill, but they produce larger quantities of cytokines and can kill more efficiently if they are activated. Several signals have been identified that can activate natural killer cells, and each of these signals is generated only when the body is under attack. For example, during a viral attack, NK cells can be activated by IFN-α or IFN-β given off by other immune system cells. Or if bacteria invade, NK cells can be activated when their surface receptors detect the bacterial cell membrane component LPS.
Natural killer cells can destroy some tumor cells, virus-infected cells, bacteria, parasites, and fungi. They kill these cells by forcing them to commit suicide. In some cases, NK cells employ an "injection system" that uses perforin proteins to help deliver "suicide" enzymes (e.g., granzyme B) into a target cell. In other situations, a protein called Fas ligand on the NK cell surface interacts with a protein called Fas on the surface of its target, signaling the target cell to self-destruct.
The method NK cells employ to identify their targets is quite different from that of killer T cells. Natural killer cells do not have T cell receptors – the receptors that are constructed by mixing and matching gene segments. The surface receptors that natural killer cells use for target recognition are of two types: activating receptors which, when engaged, motivate the NK cell to kill, and inhibitory receptors which, when engaged, encourage it not to kill.
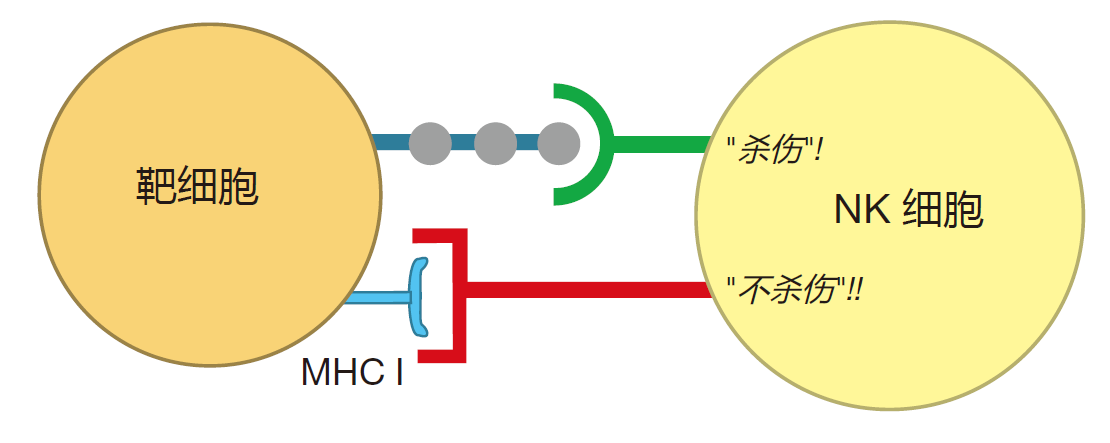
The "don't kill" signal is conveyed by inhibitory receptors that recognize class I MHC molecules on the surface of a potential target cell. Class I MHC molecules are found in varying amounts on the surface of most healthy cells in our bodies. Consequently, the presence of this surface molecule is an indication that a cell is doing okay. In contrast, "kill" signals involve interactions between the activating receptors on the surface of an NK cell and unusual carbohydrates or proteins on the surface of a target cell. These peculiar surface molecules act as flags which indicate that the target cell has been "stressed," usually because it has been infected by a virus or is becoming cancerous. Natural killer cells "measure" the relative strengths of the "kill" and "don't kill" signals to evaluate the health of a cell, and to determine whether that cell should be destroyed.
Now, why do you think it would be a good idea to have NK cells destroy cells that do not express class I MHC molecules? You remember that by examining peptides displayed by class I MHC proteins, killer T cells are able to "look inside" cells to see if anything is wrong. But what if some clever virus were to turn off expression of MHC molecules in the cells it infects? Wouldn't those virus-infected cells then be "invisible" to killer T cells? Indeed they would be. So, in those cases, it would be great to have another weapon that could kill the virus-infected cells that don't display MHC molecules on their surface. And that's just what natural killer cells can do.
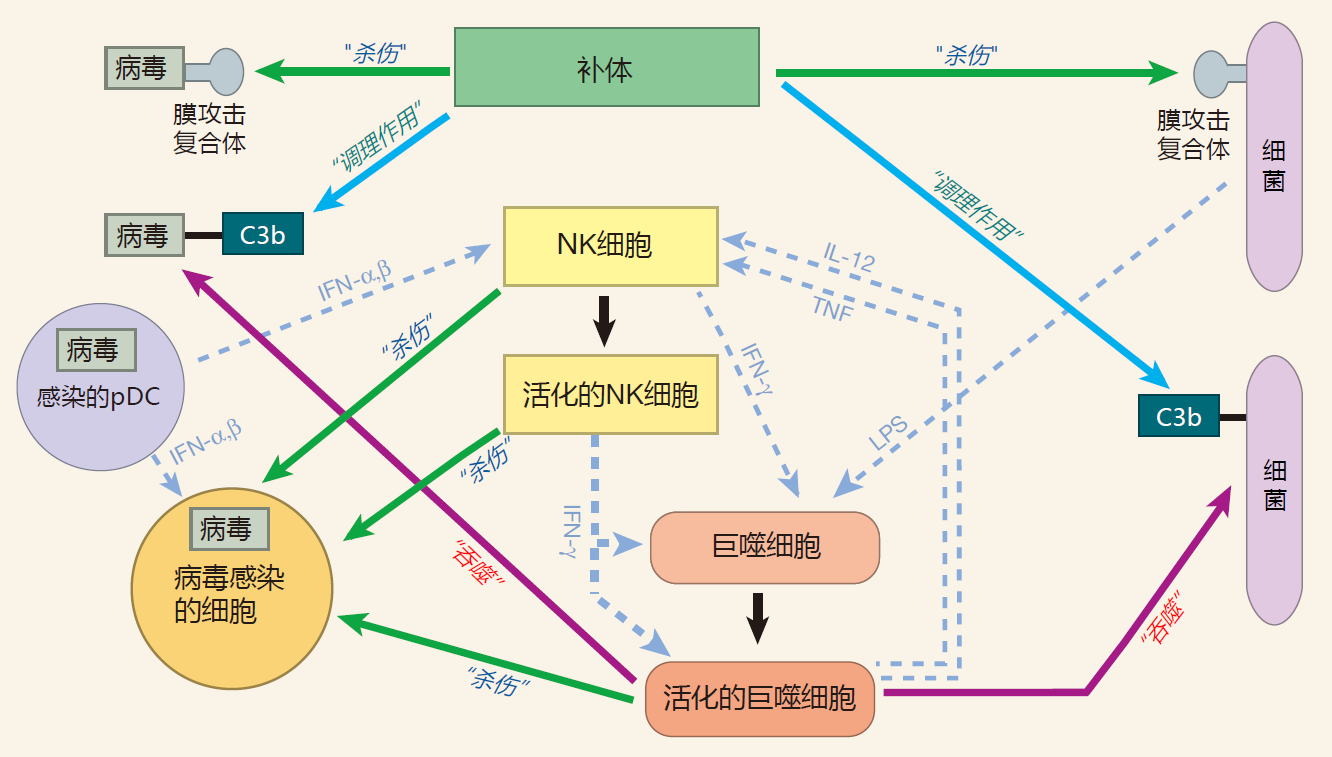
THE INNATE IMMUNE SYSTEM – A COOPERATIVE EFFORT
To make the innate system work efficiently, there must be cooperation between players. For example, neutrophils are on call from the blood. And who does the calling? The sentinel cell, the macrophage. So here we have a defense strategy in which "garbage collectors" alert the "hired guns" when their help is needed. Indeed, cooperation between macrophages and neutrophils is essential for mounting an effective defense against invading microbes. Without macrophages to summon them to sites of attack, neutrophils would just go around and around in the blood. And without neutrophils to help them, macrophages would be hard pressed to deal with sizable infections.
Also, during a bacterial infection, molecules such as LPS bind to receptors on the surface of natural killer cells, signaling that an attack is on. NK cells then respond by producing significant amounts of IFN-γ.
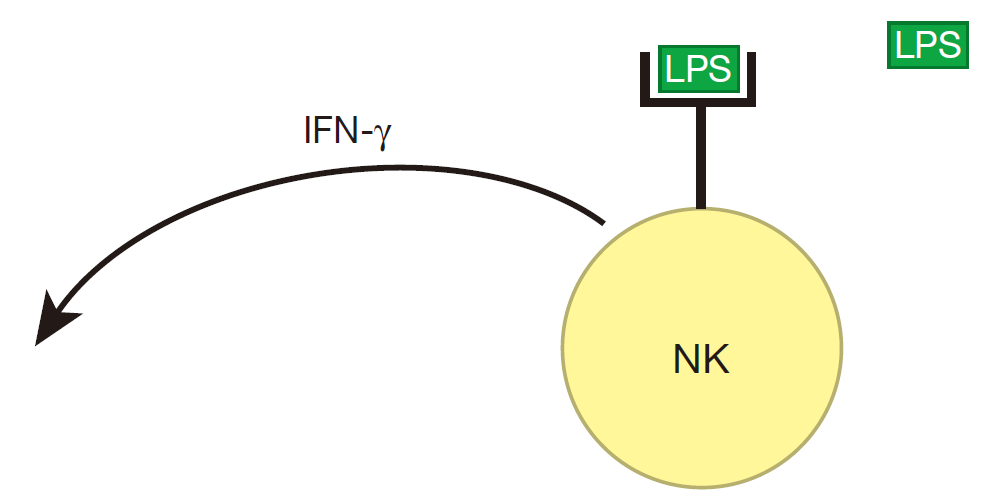
The IFN-γ produced by NK cells can prime macrophages (MΦ), which can then be hyperactivated when their receptors also bind to LPS.
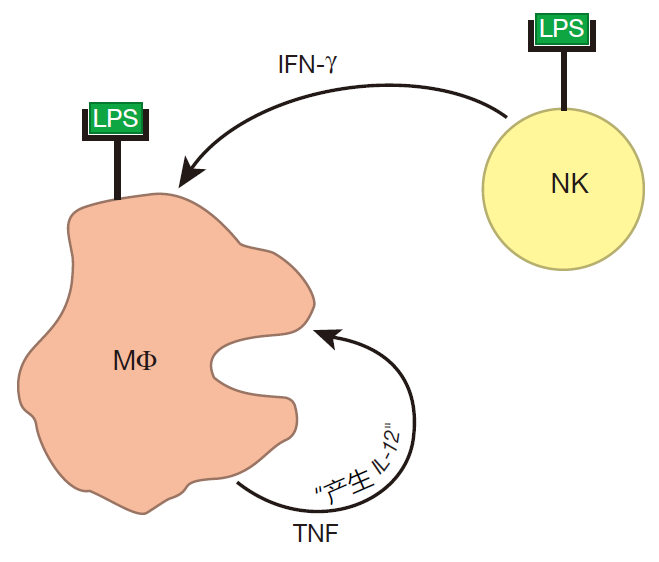
When a macrophage is hyperactivated, it produces lots of TNF. Importantly, a macrophage has receptors on its surface to which this cytokine can bind, so a macrophage can respond to the TNF it produces. And when TNF binds to these receptors, the macrophage begins to secrete IL-12. Together, TNF and IL-12 influence NK cells to increase the amount of IFN-γ they produce. And when there is more IFN-γ around, more macrophages can be primed.
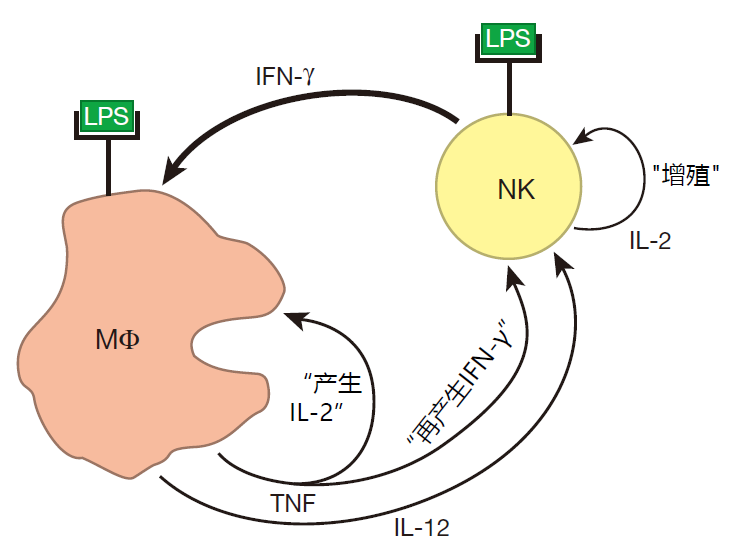
There is something else interesting going on here. IL-2 is a growth factor that is produced by NK cells. Normally, NK cells don't express the receptor for IL-2, so they don't proliferate in response to this cytokine – even though they are making it. During an infection, however, the TNF produced by macrophages upregulates the expression of IL-2 receptors on the surface of NK cells. Consequently, NK cells can now react to the IL-2 they make and begin to proliferate. As a result of this proliferation, there will soon be many more NK cells to defend against an invasion – and to help activate more macrophages. So macrophages and NK cells cooperate in several different ways to strengthen the response of the innate system to an attack.
Professional phagocytes and the complement system also work together. As we discussed, complement protein fragments such as iC3b can tag invaders for phagocyte ingestion. But complement opsonization also can play a role in activating macrophages. When C3 fragments that are decorating an invader bind to receptors on the surface of a macrophage, this provides an activation signal for the macrophage which is similar to that supplied by LPS. This is a good idea because there are invaders that can be opsonized by complement but which do not make LPS. Cooperation between the complement system and the phagocytes is not a one-way street. Activated macrophages actually produce several of the most important complement proteins: C3, factor B, and factor D. So in the heat of battle, when complement proteins may be depleted out in the tissues, macrophages can help resupply the complement system. In addition, during an infection, macrophages secrete chemicals that increase the permeability of nearby blood vessels. And when these vessels become leaky, more complement proteins are released into the tissues.
These interactions between phagocytes, NK cells, and the complement proteins are examples of the many ways in which innate system players work together. Only by cooperating with each other can the players on the innate system team respond quickly and strongly to an invasion.
A PROPORTIONAL RESPONSE
In reacting to an attack, our military tries to mount a response that is proportional to the threat. Such a proportional response insures that, on the one hand, resources will not be wasted by overreacting and, on the other hand, that the reaction will be strong enough to get the job done. The immune system is also set up to provide a proportional response to microbial invasions. For example, the number of macrophages engaged in battle depends on the size of the attack and the amount of chemicals given off by macrophages to summon neutrophils or activate NK cells depends on how many macrophages are fighting. Consequently, the more serious the invasion, the more macrophages will be involved, and the more neutrophils and NK cells will be mobilized. Likewise, the larger a bacterial invasion is, the more "danger molecules" such as LPS will be present at the battle scene. And the more LPS there is, the more NK cells will be activated to produce battle cytokines such as IFN-γ – which help activate macrophages. Because the magnitude of the immune response is directly linked to the seriousness of the attack, "the punishment usually fits the crime."
REVIEW
Complement proteins participate in the construction of membrane attack complexes that can puncture and destroy some bacteria and viruses. Complement proteins can also tag pathogens for ingestion by professional phagocytes, and can act as chemoattractants to recruit phagocytic cells to the battle site.
Complement proteins are present in high concentrations in the blood and in the tissues, so they are always ready to go. This is one of the most important features of the complement system: It works really fast. However, for the complement system to spring into action, it must first be activated. Activation by the alternative (spontaneous) pathway simply requires that a complement protein fragment, C3b, bind to an amino or hydroxyl group on an invader. Because these chemical groups are ubiquitous, the default option in this system is death: Any surface that is not protected against binding by complement fragments will be targeted for destruction. Fortunately, there are multiple mechanisms which protect human cells from complement attack.
In addition to the alternative activation pathway, which can be visualized as grenades going off randomly here and there, there is a second pathway for activating the complement system that is more directed: the lectin activation pathway. Here, a protein called mannose-binding lectin acts as a "guidance system" which targets the complement "bombs" to invaders that have distinctive carbohydrate molecules on their surfaces.
Macrophages and neutrophils are professional phagocytes. Long-lived macrophages reside beneath the surface of all the parts of our body that are exposed to the outside world. There these phagocytic cells act as sentinels. Most of the time, macrophages just eat dead cells and debris. However, if they find an invader, they become activated. In this activated state, they can present antigens to T cells, they can send out signals that recruit other immune system cells to help in the struggle, and they can become vicious killers.
In contrast to sentinel macrophages, most neutrophils can be found in the blood – where they are on call in case of attack. Whereas macrophages are quite versatile, neutrophils mainly do one thing – kill. Neutrophils use cellular adhesion molecules to exit blood vessels at sites of inflammation, and as they exit, they are activated to become killers. Fortunately, these cells only live about five days. This limits the damage they can do to healthy tissues once an invader has been vanquished. On the other hand, if the attack is prolonged, there are plenty more neutrophils that can exit the blood and help out.
Cells of the innate immune system are equipped with pattern-recognition receptors that detect signatures of whole classes of commonly encountered bacteria and viruses. Some PRRs also recognize signals given off by dying cells. When these danger signals are detected, sentinel cells such as macrophages respond by producing battle cytokines that alert other cells and prepare them to repulse the attack.
In response to a viral infection, the pattern-recognition receptors of most cells in the body can trigger the production of type I interferons, IFN-α or IFN-β. These proteins can bind to interferon receptors on the cells that produce them, and this binding results in the expression of hundreds of genes that can limit the virus's ability to reproduce within the infected cells. IFN-α and IFN-β can also function as warning proteins. When they bind to IFN receptors on nearby uninfected cells, they prepare these cells for a viral attack. The warned cells not only produce proteins that will hinder viral replication, but they are also prepared to commit suicide if they are attacked. This is an altruistic act because both the infected cells and the viruses within them are destroyed, limiting the spread of the virus to other cells. One of the body's sentinel cells, the plasmacytoid dendritic cell, can produce huge quantities of type I interferons when infected by a virus. For this reason, pDCs are important players in the innate immune system's defense against a viral attack.
The natural killer cell is another player on the innate team which is on call from the blood. These cells are a cross between a killer T cell (CTL) and a helper T cell. NK cells resemble helper T cells in that they secrete cytokines which affect the function of both innate and adaptive immune systems. And like CTLs, natural killer cells can destroy infected cells. However, in contrast to killer T cells, which select their targets by surveying peptides displayed by class I MHC molecules, NK cells focus on killing cells that do not express class I MHC molecules – especially stressed cells that have lost class I MHC expression due to a viral infection.
Phagocytes and the complement proteins provide an immediate response to an attack because these weapons are already in place. As the battle continues, signals given off by the innate system recruit even more defenders from the blood stream, and the innate system warriors cooperate to strengthen the defense. By working together, members of the innate system team provide a fast and effective response to common invaders. Importantly, the system is designed to elicit a defense which avoids overreaction, yet is adequate to the task.
SUMMARY FIGURE
In this figure, I have summarized some of the concepts we discussed in this lecture. For clarity, I have chosen a macrophage as a representative of the professional phagocytes, a bacterium as an example of an invader which can reproduce without entering human cells, and a virus as an example of an invader which must enter a human cell to complete its life cycle. After Lectures 3, 4, and 6, I will expand this figure to include players from the adaptive immune system as they take the field.