第3讲 B 淋巴细胞和抗体
HEADS UP!
B cells and the antibodies they produce are part of the adaptive immune system. B cells must be activated before they can make antibodies. "Fail-safe" mechanisms help prevent inappropriate B cell activation, and the principle of clonal selection insures that only those B cells which make antibodies appropriate to defend against an invader are mobilized. A "mix-and-match" scheme is used to construct the genes that encode a B cell's antibodies, and during the course of an attack, B cells can upgrade the antibodies they produce to mount a more targeted defense.
INTRODUCTION
Microbes such as bacteria and viruses are always mutating. Just as mutations in bacteria can render them resistant to certain antibiotics, mutations also can change microbes in ways that make them better able to resist immune defenses. When this happens, the immune system must "adapt" by producing new counter-weapons to keep the mutated microbe from taking over. Indeed, a chess match has been going on for millions of years in which the immune systems of animals have been constantly "upgraded" in response to novel weapons fielded by microbial attackers. The most striking upgrade of the immune system began about 200 million years ago, when, in fish, evolution led to the precursor of what might be called the "ultimate defense" – a system so adaptable that, in principle, it can protect against any possible invader. This defense, the adaptive immune system, has reached its most sophisticated form in humans. Indeed, without an immune system which can recognize and adapt to deal with unusual invaders, human life would not be possible.
In this lecture, we will focus on one of the most important components of the adaptive immune system: the B cell. Like all the other blood cells, B cells are born in the bone marrow, where they descend from stem cells. About one billion B cells are produced each day during the entire life of a human, so even old guys like me have lots of freshly made B cells. During their early days in the marrow, B cells select gene segments coding for the two proteins that make up their B cell receptors (BCRs), and these receptors then take up their positions on the surface of the B cell. The antibody molecule is almost identical to the B cell receptor except that it lacks the protein sequences at the tip of the heavy chain which anchor the BCR to the outside of the cell. Lacking this anchor, the antibody molecule is exported out of the B cell and is free to travel around the body to do its thing.
THE B CELL RECEPTOR
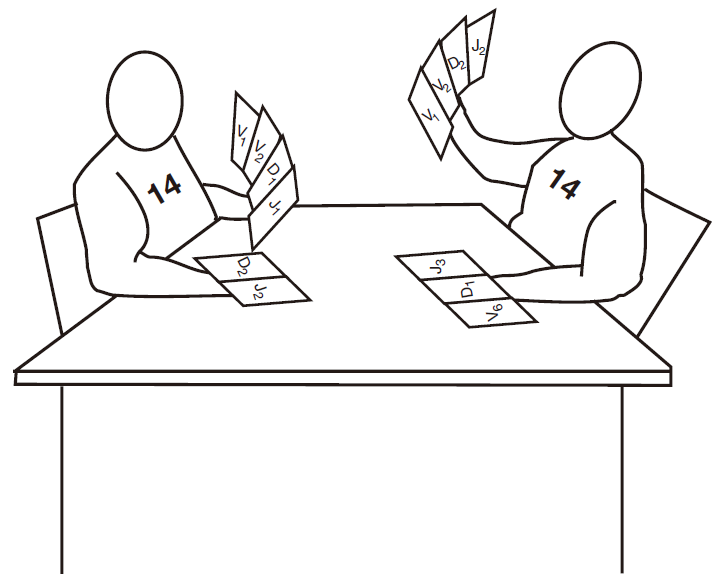
I want to tell you a little about the process of selecting gene segments to make a B cell receptor. I think you'll find it interesting – especially if you like to gamble. The BCR is made up of two kinds of proteins, the heavy chain (Hc) and the light chain (Lc), and each of these proteins is encoded by genes that are assembled from gene segments. The gene segments that will be chosen to make up the final Hc gene are located on chromosome 14, and each B cell has two chromosome 14s (one from Mom and one from Dad). This raises a bit of a problem because, as we discussed earlier, each B cell makes only one kind of antibody. Therefore, because there are two sets of Hc segments, it is necessary to "silence" the segments on one chromosome 14 to keep a B cell from making two different Hc proteins. Of course, Mother Nature could have chosen to make one chromosome a "dummy," so that the other would always be the one that was used – but she didn't. That would have been too boring! Instead, she came up with a much sweeter scheme, which I picture as a game of cards with the two chromosomes as players. It's a game of "winner takes all" in which each player tries to rearrange its cards (gene segments) until it finds an arrangement that works. The first player to do this wins.
You will remember from the first lecture that the finished heavy chain protein is assembled by pasting together four separate gene segments (V, D, J, and C), and that lined up along chromosome 14 are multiple, slightly different copies of each kind of segment.
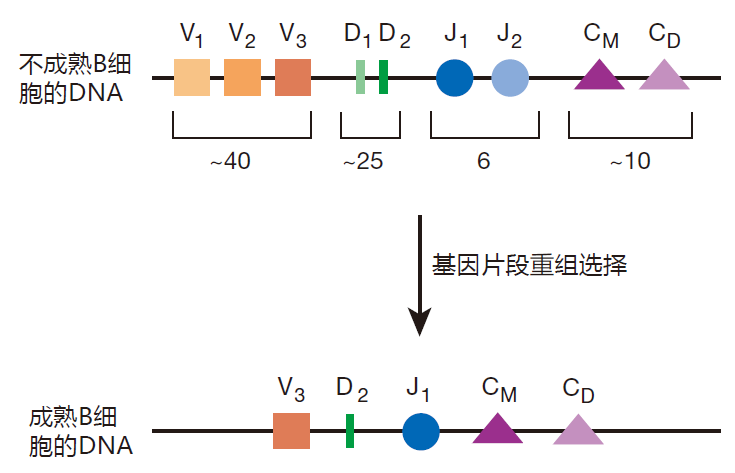
The players in this card game first choose one each of the possible D and J segments, and these are joined together by deleting the DNA sequences in between them. Then one of the many V segments is chosen, and this "card" is joined to the DJ segment, again by deleting the DNA in between. Right next to the J segment is a string of gene segments (C M, C D, etc.) that code for various constant regions. By default, the constant regions for IgM and IgD are used to make the initial BCR, just because they are first in line. Immunologists call these joined-together gene segments a "gene rearrangement," but it is really more about cutting and pasting than rearranging. Anyway, the result is that the chosen V, D, and J segments and the constant region segments all end up adjacent to each other on the chromosome.
Protein translation stops when the ribosome encounters one of the three stop codons. So if the gene segments are not joined up just right (in frame), the protein translation machinery will encounter a stop codon and terminate protein assembly somewhere in the middle of the heavy chain. If this happens, the result is a useless little piece of protein. In fact, you can calculate that each player only has about one chance in nine of assembling a winning combination of gene segments that will produce a full-length Hc protein. Immunologists call such a combination of gene segments a productive rearrangement. If one of the chromosomes that is playing this game ends up with a productive rearrangement, that chromosome is used to construct the winning Hc protein. This heavy chain protein is then transported to the cell surface, where it signals to the losing chromosome that the game is over. The details of how the signal is sent, and how it stops the rearrangement of gene segments on the other chromosome remain to be discovered. However, it is thought to have something to do with changing the conformation of the DNA on the losing chromosome – so that it is no longer accessible to the cut-and-paste machinery.
Since each player only has about a one in nine chance of success, you may be wondering what happens if both chromosomes fail to assemble gene segments that result in a productive rearrangement. Well, the B cell dies. That's right, it commits suicide! It's a high-stakes game, because a B cell that cannot express a receptor is totally useless.
If the heavy chain rearrangement is productive, the baby B cell proliferates for a bit, and then the light chain players step up to the table. The rules of their game are similar to those of the heavy chain game, but there is an additional test which must be passed to win: The completed heavy and light chain proteins must fit together properly to make a complete antibody. If the B cell fails to productively rearrange heavy and light chains, or if the Hcs and Lcs don't match up correctly, the B cell commits suicide.
The result of this contest is that although a B cell can display as many as 100,000 BCRs on its surface, every mature B cell produces one and only one kind of BCR or antibody, made up of one and only one kind of Hc and Lc. Nevertheless, because a mix-and-match strategy is used to make the final Hc and Lc genes of each B cell, the receptors on different B cells are so diverse that collectively, our B cells can probably recognize any organic molecule that could exist. When you consider how many molecules that might be, the fact that a simple scheme like this can create such diversity is truly breathtaking.
HOW THE BCR SIGNALS
Immunologists call the antigen that a given B cell's receptors recognize its cognate antigen, and the tiny region of the cognate antigen that a BCR actually binds to is called its epitope. For example, if a B cell's cognate antigen happens to be a protein on the surface of the flu virus, the epitope will be the part of that protein (usually 6–12 amino acids) to which the BCR actually binds. When the BCR recognizes the epitope for which it is matched, it must signal this recognition to the nucleus of the B cell, where genes involved in activating the B cell can be turned on or off. But how does this BCR "antenna" send a signal to the nucleus that it has found its epitope? At first sight it would appear that this could be a bit of a problem, because, as you can see from this figure, the part of the heavy chain that extends through the cell membrane into the interior of the cell is only a few amino acids in length – way too short to do any serious signaling.
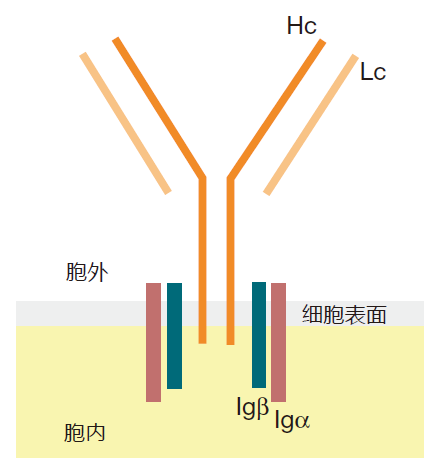
To make it possible for the external part of the BCR to signal what it has seen, B cells are equipped with two accessory proteins, Igα and Igβ, which associate with the heavy chain protein and extend into the inside of the cell. Thus, the complete B cell receptor really has two parts: the Hc/Lc part outside the cell that recognizes the antigen but can't signal, plus the Igα and Igβ proteins that can signal, but which are totally blind to what's going on outside the cell.
To generate an activation signal, many BCRs must be brought close together on the surface of the B cell. When BCRs are clustered like this, immunologists say they are crosslinked – although the receptors really are not linked together. B cell receptors can be clustered, for example, when they bind to an epitope that is present multiple times on a single antigen (e.g., a protein in which a sequence of amino acids is repeated many times).
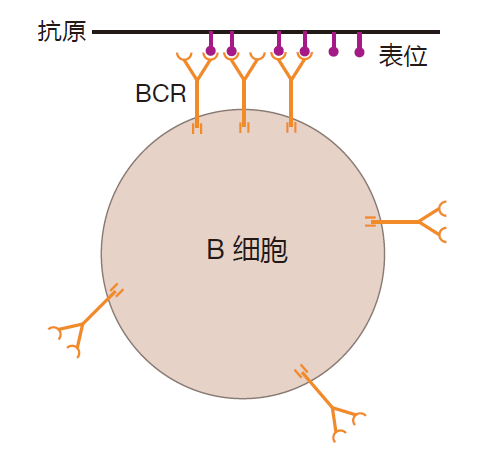
Crosslinking of BCRs also can result when BCRs bind to epitopes on individual antigens that are close together on the surface of an invader. Indeed, the surfaces of most bacteria, viruses, and parasites are composed of many copies of a few different proteins. So if a B cell's receptors recognize an epitope on one of these proteins, lots of BCRs can be clustered. Indeed, the requirement for crosslinking is one way B cells focus on common enemies. Finally, B cell receptors also can be brought together by binding to epitopes on antigens that are clumped together (e.g., a clump of proteins). Regardless of how it is accomplished, crosslinking of B cell receptors is essential for B cell activation. Here's why.
The tails of the Igα and Igβ proteins interact with signaling molecules inside the cell. And when enough of these interactions are concentrated in one region, an enzymatic chain reaction is initiated which sends a message to the nucleus of the cell saying, "BCR engaged." So the trick to sending this message is to get lots of Igα and Igβ molecules together – and that's exactly what crosslinking a B cell's receptors does. The clustering of BCRs brings enough Igα and Igβ molecules together to set off the chain reaction that sends the "BCR engaged" signal. So BCR crosslinking is key.
You remember from the last lecture that fragments of the complement proteins (e.g., C3b) can bind to (opsonize) invaders. This tag indicates that the invader has been recognized as dangerous by the innate immune system, and invites innate system players such as macrophages to destroy the opsonized invader. It turns out that antigens opsonized by complement fragments also can alert the adaptive immune system. Here's how.
In addition to the B cell receptor and its associated signaling molecules, there is another protein on the surface of a B cell that can play an important role in signaling. This protein is a receptor that can bind to complement fragments which are decorating an invader. Consequently, for an opsonized antigen, there are two receptors on a B cell that can bind to the antigen: the BCR, which recognizes a specific epitope on the antigen, and the complement receptor that recognizes the "decorations." When this happens, the opsonized antigen acts as a "clamp" that brings the BCR and the complement receptor together on the surface of the B cell.
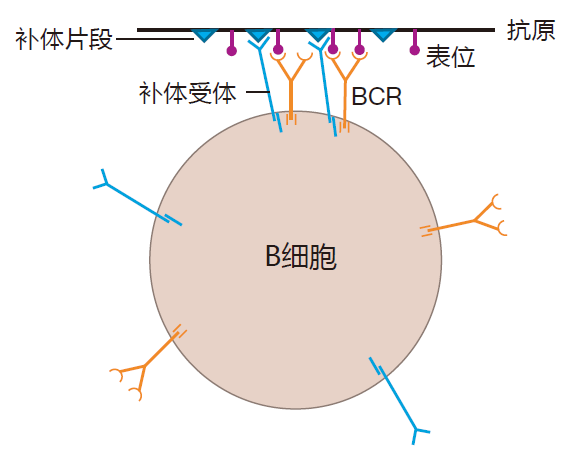
When the BCR and the complement receptor are brought together in this way by opsonized antigen, the signal that the BCR sends is greatly amplified. What this means in practice is that the number of BCRs that must be clustered to send the "receptor engaged" signal to the nucleus is decreased at least 100-fold. Because the complement receptor can have such a dramatic effect on signaling, it is called a co-receptor. The function of this co-receptor is especially important during the initial stages of an attack, when the amount of antigen available to crosslink B cell receptors is limited. Recognition of opsonized invaders by the B cell's co-receptor serves to make B cells exquisitely sensitive to antigens which the innate system already has identified as dangerous. This is an excellent example of the "instructive" function of the innate system. Indeed, the decision on whether an invader is dangerous or not usually is made by the innate, not the adaptive system.
HOW B CELLS ARE ACTIVATED
To produce antibodies, B cells must first be activated. B cells that have never been activated by encountering their cognate antigen are called naive or virgin B cells. An example would be a B cell that can recognize the small-pox virus, but which happens to reside in a human who has never been exposed to smallpox. In contrast, B cells that have encountered their cognate antigen and have been activated are called experienced B cells. There are two ways that naive B cells can be activated to defend against invaders. One is completely dependent on the assistance of helper T cells (T cell-dependent activation) and the second is more or less independent of T cell help (T cell-independent activation).
T cell-dependent activation
Activation of a naive B cell requires two signals. The first is the clustering of the B cell's receptors and their associated signaling molecules. However, just having its receptors crosslinked is not enough to fully activate a B cell – a second signal is required. This is called the-co-stimulatory signal. In T cell-dependent activation, this second signal is supplied by a helper T (Th) cell. The best-studied co-stimulatory signal involves direct contact between a B cell and a Th cell. On the surface of activated helper T cells are proteins called CD40L. If a B cell's receptors have been crosslinked, and if CD40L plugs into (ligates) a protein called CD40 on the surface of the B cell, that B cell will be activated.
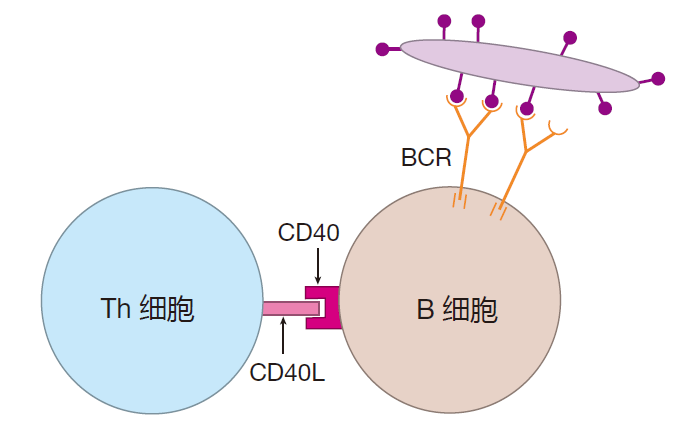
The interaction between these two proteins, CD40 and CD40L, is clearly very important for B cell activation. Humans who have a genetic defect in either of these proteins are unable to mount a T cell-dependent antibody defense.
T cell-independent activation
In response to certain antigens, virgin B cells can also be activated with little or no T cell help. This mode of activation is termed T cell-independent. What these antigens have in common is that they have repeated epitopes which can crosslink a ton of B cell receptors. A good example of such an antigen is a carbohydrate of the type found on the surface of many bacterial cells. A carbohydrate molecule is made up of many repeating units, much like beads on a string. If each "bead" is recognized by the BCR as its epitope, the string of beads can bring together many, many BCRs. The crosslinking of such a large number of BCRs can partially substitute for co-stimulation by CD40L, and can cause a B cell to proliferate. But to be fully activated and produce antibodies, a naïve B cell must receive a second signal.
This second "key" is an unambiguous "danger signal" –a clear indication that an attack is on. For example, in addition to their BCRs, B cells express Toll-like receptors, and these TLRs can alert a B cell to danger and provide the second key needed for T cell-independent activation. What is important here is that if a B cell has BCRs that can recognize a molecule with repeated epitopes such as, for example, your own DNA, it may proliferate, but fortunately, no anti-DNA antibodies will be produced. The reason is that your immune system is not engaged in a battle with your own DNA, so there will be no danger signals to provide the necessary co-stimulation. On the other hand, if the innate immune system is battling a bacterial infection, and a B cell's receptors recognize a carbohydrate antigen with repeated epitopes on the surface of the bacterial invader, that B cell will produce antibodies –because danger signals from the battleground can supply the second key needed for complete B cell activation. Of course, as is true of T cell-dependent activation, T cell-independent activation is antigen specific: Only those B cells whose receptors recognize the repeated epitope will be activated.
One advantage of T cell-independent activation is that B cells can jump right into the fray without having to wait for helper T cells to be activated. The result is a speedier antibody response. Most B cells that are activated without T cell help are found in the spleen. These "helpless" B cells can mount a rapid defense against bacteria such as Streptococcus pneumoniae by making IgM antibodies that recognize epitopes on the polysaccharide capsule that surrounds the bacterium. The importance of this T cell-independent activation is demonstrated by the fact that humans who have had their spleens removed are at high risk for infection by Streptococcus pneumoniae and other encapsulated bacteria.
There is something else important going on here. Helper T cells only recognize protein antigens – the peptides presented by class II MHC molecules. Consequently, if all B cell activation required T cell help, the entire adaptive immune system would be focused on proteins. This wouldn't be so great, because many common invaders have carbohydrates or fats on their surface that are not found on the surface of human cells. Therefore, these carbohydrates and fats make excellent targets for recognition by the immune system. So allowing some antigens to activate B cells without T cell help is a wonderful thing: It increases the universe of antigens that the adaptive immune system can react against to include not only proteins, but carbohydrates and fats as well.
The logic of B cell activation
You may be asking yourself: Why does B cell activation require two signals? Wouldn't things go more quickly if all a B cell needed for activation was crosslinking of its receptors? Yes, this probably would speed up antibody production, but it would also be way too dangerous. Because of the diversity of B cell receptors, there is essentially no limit on what they can recognize – including our own proteins, carbohydrates, and fats. Most B cells which can recognize our own molecules are eliminated shortly after they are born in the bone marrow (much more on this in Lecture 9). However, this screening process is not 100% effective, and there are self-reactive B cells in circulation which could cause autoimmune disease if they produced antibodies (autoantibodies). To guard against this possibility there is a fail-safe mechanism in place which allows B cell activation only when there is real danger. That's where the second signal comes in. For T cell-dependent activation, the B cell and the Th cell must agree that there is a threat before the B cell can receive this second signal. For T cell-independent activation, the second signal is a clear indication that there has been an invasion – a dangerous situation which warrants B cell activation.
Polyclonal activation
In addition to T cell-dependent and T cell-independent activation of B cells, there is an "unnatural" way that B cells can be activated. In this case, the antigen, usually called a mitogen, binds to molecules on the B cell surface that are not B cell receptors, and brings these molecules together.
When this happens, BCRs that are associated with these molecules also can be clustered. In contrast to T cell-dependent and T cell-independent activation, this polyclonal activation does not depend on the cognate antigen recognized by the BCR – the BCR just comes along for the ride. In this way, many different B cells with many different specificities can be activated by a single mitogen. Indeed, mitogens are favorite tools of immunologists, because they can be used to activate a lot of B cells simultaneously, making it easier to study events that take place during activation.
One example of a mitogen is the highly repetitive structure that makes up the surface of certain parasites. During a parasitic infection, the molecules that make up these structures can bind to receptors (mitogen receptors) on the surface of B cells and cluster them. And when the mitogen receptors are clustered in this way, the cell's BCRs are also dragged together. The result is polyclonal activation of B cells. But why would the immune system want to react to a parasitic attack by activating B cells whose BCRs do not even recognize the parasite? The answer is that this is not something the immune system was designed to do! By activating a bunch of B cells that will produce irrelevant antibodies, the parasite seeks to distract the immune system from focusing on the job at hand – destroying the parasitic invader. So polyclonal activation of B cells by a mitogen actually is an example of the immune system gone wrong – a subject we will discuss at length in another lecture.
CLASS SWITCHING
Once B cells have been activated and have proliferated to build up their numbers, they are ready for the next stage in their life: maturation. Maturation can be divided roughly into three steps: class switching, in which a B cell can change the class of antibody it produces; somatic hypermutation, in which the rearranged genes for the B cell receptor can mutate to increase the affinity of the BCRs for their cognate antigen; and a "career decision," during which the B cell decides whether to become an antibody factory (a plasma B cell) or a memory B cell. The exact order of these maturation steps varies, and some B cells may skip one or more steps altogether.
When a virgin B cell is first activated, it produces mainly IgM antibodies – the default antibody class. B cells also can produce IgD antibodies. However, IgD antibodies represent only a tiny fraction of the circulating antibodies in a human, and it is unclear whether they actually perform any significant function in the immune defense. You remember that an antibody's class is determined by the constant (Fc) region of its heavy chain – the "tail" of the antibody molecule, if you will. Interestingly, the same heavy chain messenger RNA is used to make both IgM and IgD, but the mRNA is spliced one way to yield an M-type constant region and another way to produce a D-type constant region.
As a B cell matures, it has the opportunity to change the class of antibody it makes to one of the other antibody classes: IgG, IgE, or IgA. Located just next to the gene segment on chromosome 14 that encodes the constant region for IgM are the constant region segments for IgG, IgE, and IgA. So all that a B cell has to do to switch its class is to cut off the IgM constant region DNA and paste on one of the other constant regions (deleting the DNA in between). Special switching signals that allow this cutting and pasting to take place are located between the constant region segments. For example, here's what happens when a B cell switches from an IgM constant region (C M in this drawing) to an IgG constant region (C G ):
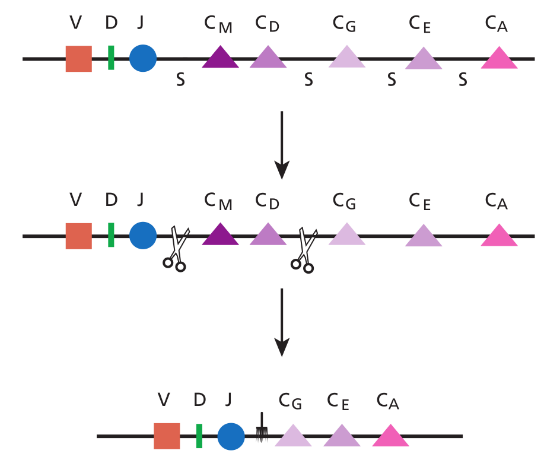
The net result of class switching is that although the part of the antibody that binds to the antigen (the Fab region) remains the same, the antibody gets a new Fc region. This is an important change, because it is the constant region that determines how the antibody will function.
ANTIBODY CLASSES AND THEIR FUNCTIONS
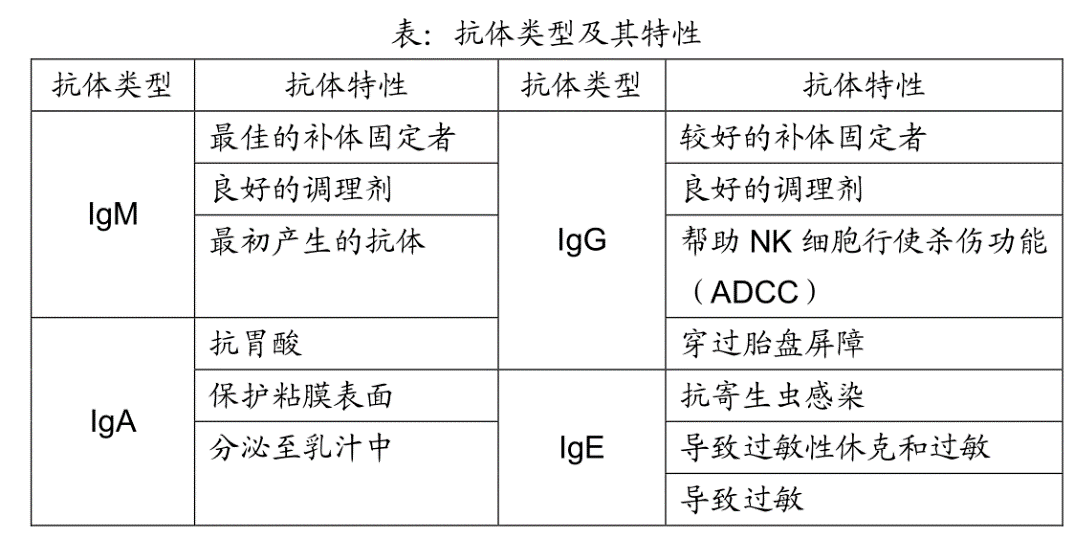
Let's take a look at the four main classes of antibodies: IgM, IgA, IgG, and IgE. As you will see, because of the unique structure of its constant region, each antibody class is particularly well suited to perform certain duties.
IgM antibodies
IgM antibodies were the first class of antibodies to evolve, and even "lower" vertebrates (my apologies to the animal rights folks) have adaptive immune systems that produce IgM antibodies. So it makes sense that in humans, when naive B cells are first activated, they make mainly IgM antibodies. You may remember that an IgG antibody looks roughly like this.
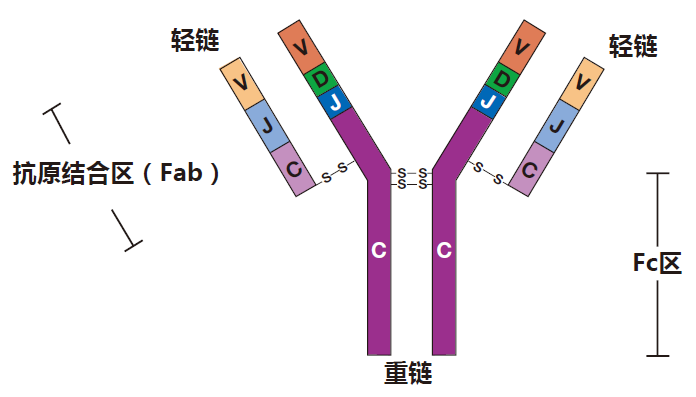
In contrast, an IgM antibody is like five IgG antibody molecules all stuck together. It's really massive!
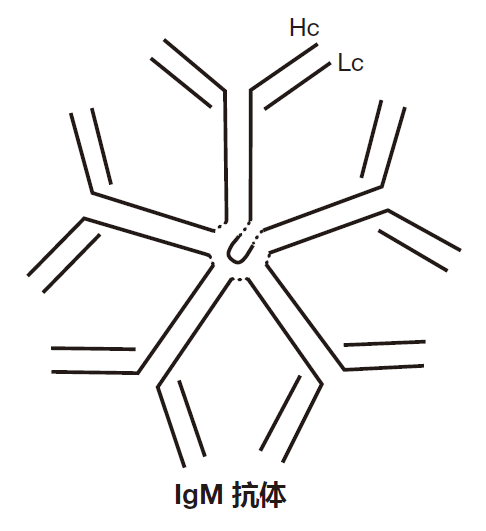
Producing IgM antibodies early during an infection actually is quite smart, because IgM antibodies are very good at activating the complement cascade (immunologists call this fixing complement). Here's how it works.
In the blood and tissues, some of the complement proteins (about 30 of them!) get together to form a big complex called C1. Despite its size, this complex of proteins cannot activate the complement cascade, because it's bound to an inhibitor molecule. However, if two or more C1 complexes are brought close together, their inhibitors fall off, and the C1 molecules can then initiate a cascade of chemical reactions that produces a C3 convertase. Once this happens, the complement system is in business because a C3 convertase converts C3 to C3b, setting up an amplification loop that produces more and more C3b. So the trick to activating the complement system by this-classical (antibody-dependent) pathway is to bring two or more of the C1 complexes together – and that's just what an IgM antibody can do.
Once the antigen-binding regions of an IgM antibody have bound to an invader, C1 complexes can bind to the Fc regions of the antibody. Because each IgM antibody has five Fc regions close together (this is the important point), two C1 complexes can bind to the Fc regions of the same IgM antibody, bringing the complexes close enough together to set off the complement cascade. So the sequence of events is: The IgM antibody binds to the invader, several C1 molecules bind to the Fc region of the IgM antibody, their inhibitors are released, and the C1 molecules trigger the complement chain reaction on the invader's surface.
The reason the classical activation pathway is so useful is that some clever bacteria have evolved coats which resist the attachment of complement proteins. Fortunately, B cells can produce antibodies which will bind to essentially any coat a bacterium might put on. Consequently, antibodies can extend the range of the complement system by helping attach complement proteins to the surface of wily bacteria. This is a nice example of the innate immune system (the complement proteins) cooperating with the adaptive immune system (IgM antibodies) to help destroy an invader. In fact, the term "complement" was coined by immunologists when they first discovered that antibodies were much more effective in dealing with invaders if they were "complemented" by other proteins – the complement proteins.
The alternative (spontaneous) complement activation pathway that we talked about in the last lecture is totally non-specific: Any unprotected surface is fair game. In contrast, the classical (antibody-dependent) activation pathway is quite specific: Only those antigens to which the antibody binds will be targeted for complement attack. In this system, the antibody identifies the invader, and the complement proteins do the dirty work.
Certain "subclasses" of IgG antibodies also can fix complement, because C1 can bind to the Fc region of these antibodies. However, IgG antibodies are real wimps, with only one Fc region per molecule. So bringing two C1 complexes close enough together to get things started requires that two molecules of IgG bind right next to each other on the surface of the invading pathogen – and this is likely to happen only when there is a lot of IgG around. So, early in an infection, when antibodies are just beginning to be made, IgM antibodies have a great advantage over IgG antibodies because they fix complement so efficiently. In addition, IgM antibodies are very good at neutralizing viruses by binding to them and preventing them from infecting cells. Because of these properties, IgM is the perfect "first antibody" to defend against viral or bacterial infections.
IgG antibodies
IgG antibodies come in a number of different subclasses that have slightly different Fc regions and, therefore, different functions. For example, one subclass of human IgG antibodies, IgG1, is very good at binding to invaders to opsonize them for ingestion by professional phagocytes.
This is because macrophages and neutrophils have receptors on their surfaces that can bind to the Fc portion of IgG1 antibodies once those antibodies have bound to an invader.
Another IgG subclass, IgG3, fixes complement better than any other IgG subclass. In addition, natural killer cells have receptors on their surface that can bind to the Fc region of IgG3 antibodies. As a result, IgG3 can form a bridge between an NK cell and its target by binding to the target cell (e.g., a virus-infected cell) with its Fab region, and to the NK cell with its Fc region. Not only does this bring the NK cell close to its target, but having its Fc receptors bound actually stimulates an NK cell to be a more effective killer. This process is called antibody-dependent cellular cytotoxicity (ADCC). In ADCC, the NK cell does the killing, but the antibody identifies the target.
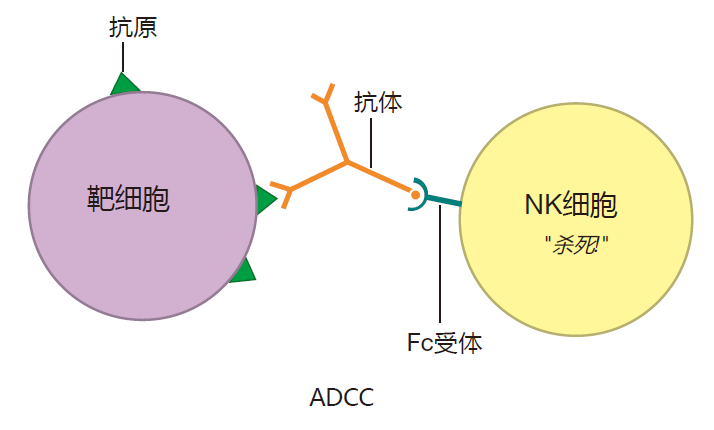
Like IgM antibodies, IgG antibodies also are very good at neutralizing viruses. Moreover, IgG antibodies are unique in that they can pass from the mother's blood into the blood of the fetus by way of the placenta. This provides the fetus with a supply of IgG antibodies to tide it over until it begins to produce its own – several months after birth. This extended protection is possible because IgG antibodies are the longest-lived antibody class, with a half-life of about three weeks. In contrast, IgM antibodies have a half-life of only about one day.
The "G" in IgG stands for "gamma," and IgG antibodies are sometimes called gamma globulins. If there is a possibility that you have been exposed to an infectious agent, say hepatitis A virus, your doctor may recommend that you get a gamma globulin shot. These shots are prepared by pooling together antibodies from a large number of people, at least some of whom have been infected with hepatitis A virus – and therefore are making antibodies against the virus. The hope is that these "borrowed" antibodies will neutralize most of the virus to which you have been exposed, and that this treatment will help keep the viral infection under control until your own immune system can be activated.
IgA antibodies
Here's a question for you: What is the most abundant antibody class in the human body? No, it's not IgG. It's IgA. This is really a trick question, because I told you earlier that IgG was the most abundant antibody class in the blood – which is true. It turns out, however, that we humans synthesize more IgA antibodies than all the other antibody classes combined. Why so much IgA? Because IgA is the main antibody class that guards the mucosal surfaces of the body, and a human has about 400 square meters of mucosal surfaces to defend. These include the digestive, respiratory, and reproductive tracts. So although there aren't a lot of IgA antibodies circulating in the blood, there are tons of them protecting the mucosal surfaces. Indeed, about 80% of the B cells that are located beneath these surfaces produce IgA antibodies.
One reason IgA antibodies are so good at defending against invaders that would like to penetrate the mucosal barrier is that each IgA molecule is rather like two IgG molecules held together by a "clip."
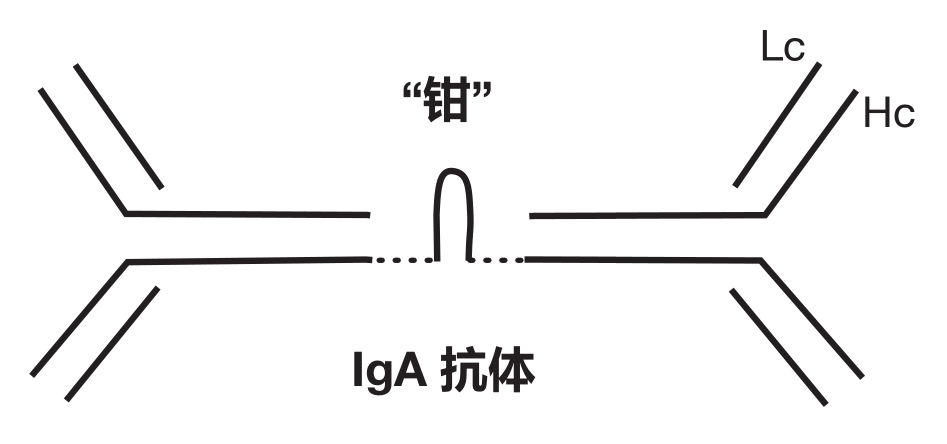
The clipped-together tail structure of IgA antibodies is responsible for several important properties of this antibody class. This structure functions as a "passport" that facilitates the transport of IgA antibodies across the intestinal wall and out into the intestine. Moreover, this unique structure makes IgA antibodies resistant to acids and enzymes found in the digestive tract. Once inside the intestine, IgA antibodies can "coat" invading pathogens and keep them from attaching to the intestinal cells they would like to infect. In addition, whereas each IgG molecule has two antigen-binding regions, the "dimeric" IgA molecule has four Fab regions to bind antigens. Consequently, dimeric IgA antibodies are very good at collecting pathogens together into clumps that are large enough to be swept out of the body with mucus or feces. In fact, rejected bacteria make up about 30% of normal fecal matter.
All together, these qualities make IgA antibodies perfect for guarding mucosal surfaces. Indeed, it is the IgA class of antibodies that is secreted into the milk of nursing mothers. These IgA antibodies coat the baby's intestinal mucosa and provide protection against pathogens that the baby ingests. This makes sense, because many of the microbes that babies encounter are taken in through their mouths – babies like to put their mouths on everything, you know.
Although IgA antibodies are very effective against mucosal invaders, they are totally useless at fixing complement: C1 won't even bind to an IgA antibody's Fc region. Again we see that the constant region of an antibody determines both its class and its function. This lack of complement-fixing activity is actually a good thing. If IgA antibodies could initiate the complement reaction, our mucosal surfaces would be in a constant state of inflammation in response to both pathogenic and non-pathogenic microbes. And, of course, having chronically inflamed intestines would not be all that great. So IgA antibodies mainly function as "passive" antibodies which block the attachment of invaders to cells that line our mucosal surfaces and usher these unwanted guests out of the body.
IgE antibodies
The IgE antibody class has an interesting history. In the early 1900s, a French physician named Charles Richet was sailing with Prince Albert of Monaco (Grace Kelly's father-in-law). The prince remarked to Richet that it was very strange how some people react violently to the toxin in the sting of the Portuguese man-of-war, and that this phenomenon might be worthy of study.
Richet took his advice, and when he returned to Paris he decided, as a first experiment, to test how much toxin was required to kill a dog. Don't ask me why he decided to use dogs in his experiments. Maybe there were a lot of stray dogs around back then, or perhaps he just didn't like working with mice. Anyway, the experiment was a success and he was able to determine the amount of toxin that was lethal. However, many of the dogs he used in this first experiment survived, because they didn't receive the lethal dose. Not being one to waste a good dog, Richet decided to inject these "leftovers" with the toxin again to see what would happen. His expectation was that these animals might have become immune to the effects of the toxin, and that the first injection would have provided protection (prophylaxis) against a second injection. You can imagine his surprise then, when all the dogs died – even the ones that received tiny amounts of toxin in the second injection. Since the first injection had the opposite effect of protection, Richet coined the word anaphylaxis to describe this phenomenon ("ana" is a prefix meaning "opposite"). Richet continued these studies on anaphylactic shock, and in 1913 he received the Nobel Prize for his work. I guess one lesson from this is that if a prince suggests you should study something, you might want to take his advice seriously!
Immunologists now know that anaphylactic shock is caused by mast cells degranulating. Like macrophages, mast cells are white blood cells that are stationed beneath all exposed surfaces (e.g., beneath the skin or the mucosal barrier). As blood cells go, mast cells are very long-lived: They can survive for years in our tissues. There they lie in wait to protect us against infection by parasites that have penetrated our barrier defenses. Stored safely inside mast cells are "granules" that contain all kinds of pre-activated, pharmacologically active chemicals, the most famous of which is histamine. Indeed, a mast cell is so full of these granules that its name is derived from the German word mastung, which means "well fed." When a mast cell encounters a parasite, it exports its granules (i.e., it "degranulates"), and dumps the contents of these granules onto the parasite to kill it.
Unfortunately, in addition to killing parasites, mast cell degranulation also can cause an allergic reaction and, in extreme cases, anaphylactic shock. Here's how this works.
An antigen (e.g., the man-of-war toxin) that can cause an allergic reaction is called an allergen. On the first exposure to an allergen, some people, for reasons that are far from clear, make a lot of IgE antibodies directed against the allergen. Mast cells have receptors (IgE receptors) on their surface that can bind to the Fc region of these IgE antibodies. And when this happens, the mast cells are like bombs waiting to explode.
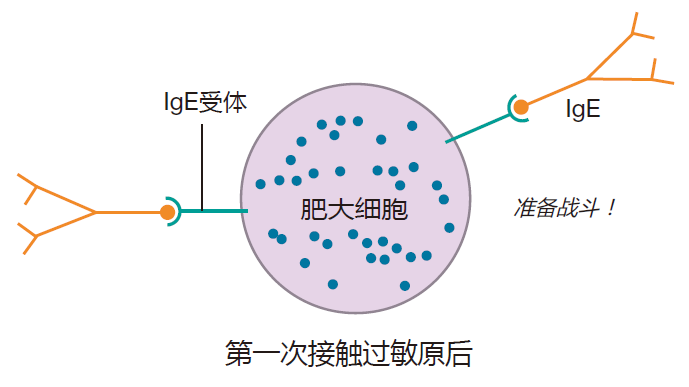
On a second exposure to the allergen, IgE antibodies that are already bound to the surfaces of mast cells can bind to the allergen. Because allergens usually are proteins with a repeating sequence, the allergen can crosslink many IgE molecules on the mast cell surface, dragging the IgE receptors together. This clustering of IgE receptors is similar to the crosslinking of B cell receptors in that bringing many of these receptors together results in a signal being sent. In this case, however, the signal says "degranulate," and the mast cell responds by dumping its granules into the surrounding tissues.
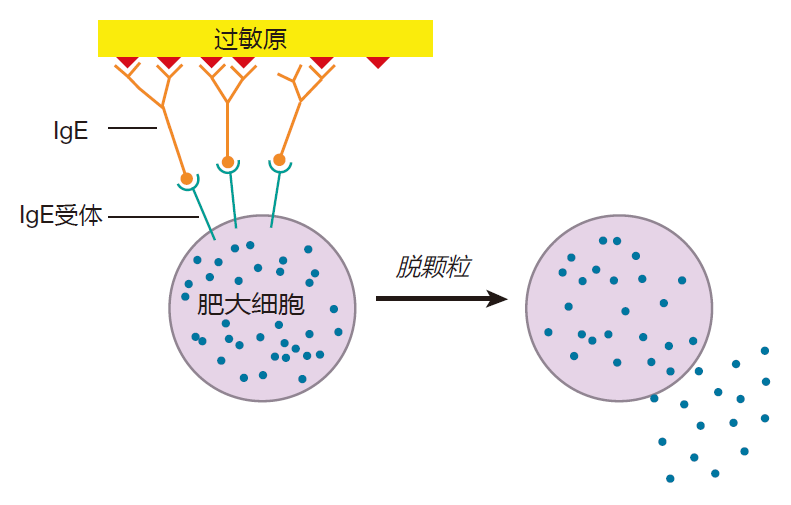
Histamines and other chemicals released from mast cell granules increase capillary permeability so that fluid escapes from the capillaries into the tissue – that's why you get a runny nose and watery eyes when you have an allergic reaction. This is usually a rather local effect, but if the toxin spreads throughout the body and triggers massive degranulation of mast cells, things can get serious. In such a case, the release of fluid from the blood into the tissues can reduce the blood volume so much that the heart no longer can pump efficiently, resulting in a heart attack. In addition, histamine from the granules can cause smooth muscles around the windpipe to contract, making it difficult to breathe. In extreme cases, this contraction can be strong enough to cause suffocation. Most of us don't have to worry about being stung by a Portuguese man-of-war. However, some people make lots of IgE antibodies in response to bee toxin, and for those folks, a bee sting can be fatal. Indeed, about 1,500 Americans die each year from anaphylactic shock.
This brings us to an interesting question: Why are B cells allowed to switch the class of antibody they make anyway? Wouldn't it be safer just to stick with good old IgM antibodies? I don't think so. Let's suppose you have a viral infection of your respiratory tract, resulting in the common cold. Would you want to be stuck making only IgM antibodies? Certainly not. You'd want a lot of IgA antibodies to be secreted into the mucus that lines your respiratory tract to bind up that virus, and help remove it from your body. On the other hand, if you have a parasitic infection (worms, for example), you'd want IgE antibodies to be produced, because IgE antibodies can cause cells such as mast cells to degranulate, making life miserable for those worms. So the beauty of this system is that the different classes of antibodies are uniquely suited to defend against different invaders.
Now suppose it could be arranged so that your immune system makes IgG antibodies when your big toe is infected, IgA antibodies when you have a cold, or IgE antibodies when you have a parasitic infection? Wouldn't that be elegant? Well, it turns out that this is exactly what happens! Here's how it works.
Antibody class switching is controlled by the cytokines that B cells encounter when switching takes place: Certain cytokines or combinations of cytokines influence B cells to switch to one class or another. For example, if B cells class switch in an environment that is rich in IL-4 and IL-5, they preferentially switch their class from IgM to IgE – just right for those worms. On the other hand, if there is a lot of IFN-γ around, B cells switch to produce IgG3 antibodies that are very effective against bacteria and viruses. Or, if a cytokine called TGFβ is present during the class switch, B cells preferentially change from IgM to IgA antibody production – perfect for the common cold. So, to insure that the antibody response will be appropriate for a given invader, all that is required is for the right cytokines to be present when B cells switch classes. But how could this be accomplished?
You remember that helper T cells are "quarterback" cells which direct the immune response. One way they do this is by producing cytokines which influence B cells to make the antibody class that is right to defend against a given invader. To learn how Th cells know which cytokines to make, you'll have to wait for the next three lectures when we discuss antigen presentation and T cell activation and function. But for now, I'll just give you the bottom line: In response to cytokines made by Th cells, B cells can switch from making IgM antibodies to producing one of the other antibody classes. As a result, the adaptive immune system can respond with antibodies tailor-made for each kind of invader – be it a bacterium, a flu virus, or a worm. What could be better than that?!
SOMATIC HYPERMUTATION
As if class switching weren't great enough, there is yet another amazing thing that can happen to B cells as they mature. Normally, the overall mutation rate of DNA in human cells is extremely low – only about one mutated base per 100 million bases per DNA replication cycle. It has to be this low or we'd all end up looking like Star Wars characters with three eyes and six ears. However, in very restricted regions of the chromosomes of B cells – those regions that contain the V, D, and J gene segments – an extremely high rate of mutation can take place. In fact, mutation rates as high as one mutated base per 1,000 bases per generation have been measured. We're talking serious mutations here! This high rate of mutation is called somatic hypermutation, and it occurs after the V, D, and J segments have been selected – and usually after class switching has taken place. So somatic hypermutation is a relatively late event in the maturation of B cells. Indeed, B cells that still make IgM antibodies usually have not undergone somatic hypermutation.
What somatic hypermutation does is to change (mutate) the part of the rearranged antibody gene that encodes the antigen-binding region of the antibody. Depending on the mutation, there are three possible outcomes: The affinity of the antibody molecule for its cognate antigen may remain unchanged, it may be increased, or it may be decreased. Now comes the neat part. In order for maturing B cells to proliferate, they must continually be re-stimulated by helper T cells. Those B cells whose BCRs have mutated to higher affinity compete more successfully for limited T cell help. Consequently, they proliferate more frequently than do B cells with lower affinity receptors. Therefore, the result of somatic hypermutation is that you end up with many more B cells whose BCRs bind tightly to their cognate antigen.
By using somatic hypermutation to make changes in the antigen-binding region of a BCR, and by using binding and proliferation to favor B cells whose mutations have increased the BCR's affinity for antigen, B cell receptors can be "fine-tuned." The result is a collection of B cells whose receptors have a higher average affinity for their cognate antigen. This whole process is called affinity maturation.
So B cells can change their constant (Fc) region by class switching and their antigen-binding (Fab) region by somatic hypermutation – and these two modifications produce B cells that are better adapted to deal with invaders. The assistance of helper T cells usually is required for B cells to make either of these upgrades. As a result, B cells that are activated without T cell help (e.g., in response to carbohydrates on the surface of a bacterium) generally don't undergo either class switching or somatic hypermutation.
B CELLS MAKE A CAREER CHOICE
The final step in the maturation of a B cell is the choice of profession. This can't be too tough, because a B cell really only has two fates to choose between: to become a plasma B cell or a memory B cell. Plasma B cells are antibody factories. If a B cell decides to become a plasma cell, it usually travels to the spleen or back to the bone marrow, and begins to produce the secreted form of the BCR – the antibody molecule. Some plasma B cells can synthesize 2,000 of these antibodies each second. However, as a result of this heroic effort, these plasma B cells only live for a few days. The ability of plasma B cells to make so many antibody molecules helps the immune system keep up with invaders such as bacteria and viruses which multiply very quickly.
Although the B cell's other possible career choice – to become a memory B cell – may not be quite so dramatic as the decision to become a plasma cell, it is extremely important. It is the memory B cell that recalls your first exposure to a pathogen and helps defend you against subsequent exposures. Immunologists haven't figured out how a B cell "chooses" to become either a memory cell or a plasma cell. However, they do know that the interaction between the co-stimulatory molecule CD40L on the surface of a helper T cell and CD40 on the B cell surface is important in memory cell generation. Indeed, memory B cells are not produced when B cells are activated without T cell help.
REVIEW
A B cell's receptors function as the "eyes" of the cell, and actually have two parts: a recognition part (made up of the heavy and light chain proteins), and a signaling part (made up of two other proteins, Igα and Igβ). The final genes that encode the recognition part are made by mixing and matching gene segments. The result is a collection of B cells with receptors so diverse that they probably can recognize any organic molecule in the universe. For these receptors to signal what they have seen requires that multiple BCRs be clustered (crosslinked). This crosslinking brings the Igα and Igβ signaling molecules that are associated with the heavy chains close together. And when enough Igα and Igβ molecules are clustered in this way, the "receptor engaged" signal is sent to the nucleus of the B cell.
B cells also have co-receptor molecules on their surface which can recognize opsonized antigen. When both the B cell's receptors and the co-receptors are engaged by an antigen, the number of BCRs which must be crosslinked to signal activation is dramatically reduced. Consequently, these co-receptors focus the attention of B cells on antigens that have already been recognized by the innate system as dangerous and which have been opsonized.
Activation of a virgin B cell requires two "keys." Crosslinking of the B cell's receptors is the first key, but a second, "co-stimulatory" key also is required. This key usually is provided by a helper T cell, and involves cell–cell contact, during which CD40L molecules on the surface of a helper T cell bind to CD40 proteins on the surface of a B cell. B cells can also be activated without T cell help. The first requirement for this T cell-independent activation is that a large number of the B cell's receptors must be crosslinked. This typically happens when the surface of an invader is made up of many copies of the antigen to which a B cell's receptors bind (its cognate antigen).
Although the crosslinking of many B cell receptors is a requisite for T cell-independent activation of a naive B cell, it is not enough. A second, co-stimulatory signal also is needed. This co-stimulation is in the form of a "danger signal" which confirms that an authentic threat exists. By demanding that two keys must be supplied before a B cell can be activated, a fail-safe system is established that guards against inappropriate B cell activation.
IgM antibodies are the first antibodies produced by B cells in response to a pathogen that has not been encountered before. However, as a B cell matures, it can choose to produce a different class of antibody: either IgG, IgA, or IgE. This class switching does not change the antigen-binding (Fab) region of the antibody. Consequently, the antibody recognizes the same antigen before and after its class has been switched.
What does change during class switching is the Fc region of the heavy chain. This is the part of the molecule that determines how the antibody functions, with some functions being better suited to certain invaders than to others. Importantly, the choice of antibody class is determined by the cytokines present in the local environment of the B cell when class switching takes place. So by arranging to have the appropriate cytokines produced at the appropriate places, the right class of antibody to defend against a particular invader can be made.
The other change that can take place as a B cell matures is somatic hypermutation. In contrast to class switching, in which the antibody gets a different Fc region, somatic hypermutation alters the antigen- binding region of the antibody. Because the probability that a B cell will proliferate depends on the affinity of its BCR for antigen, the B cells which proliferate most will be those for which somatic hypermutation has increased the binding affinity of their BCRs. The result is a collection of B cells whose BCRs, on average, bind more tightly to the invader than did the original, unmutated BCRs. These upgraded B cells are especially useful as memory cells. Because their affinity-matured BCRs are sensitive to small amounts of antigen, these B cells can be reactivated early in a second infection while the number of invaders is still relatively small.
B cells can be activated with or without T cell help, but the outcomes in these two cases usually are very different. T cell-independent activation generally results in the production of IgM antibodies. In contrast, T cell- dependent activation can result in affinity-matured IgG, IgA, or IgE antibodies. One reason for this difference is that both class switching and somatic hypermutation require ligation of CD40 proteins on B cells. This signal is usually provided by CD40L, a protein found on the surface of activated helper T cells.
As B cells mature, they must decide whether to become short-lived plasma cells, which produce vast quantities of antibodies, or to stick around as longer lived, memory B cells. These memory B cells are responsible for making the antibodies which can protect us from a subsequent attack by the same pathogen.
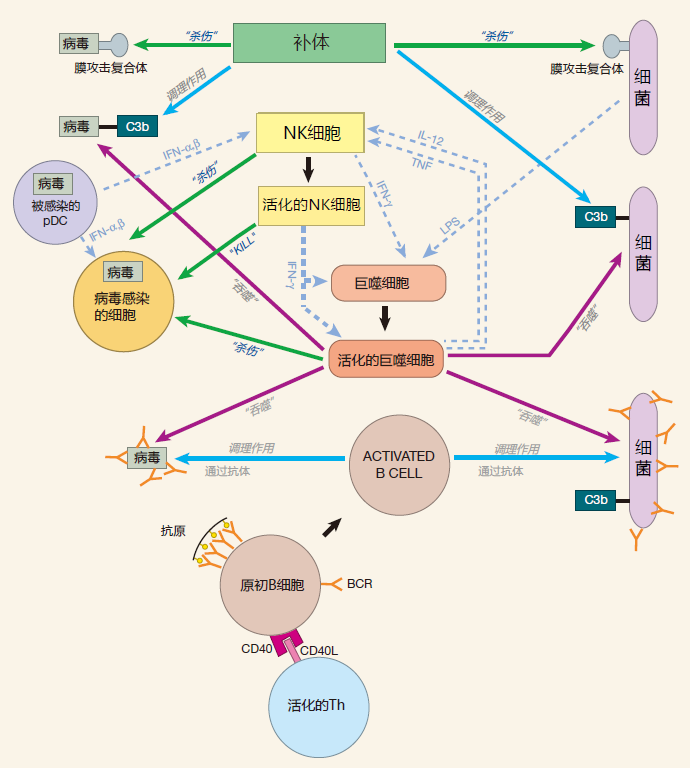
SUMMARY FIGURE
Our summary figure now includes the innate immune system from the last lecture plus B cells and antibodies.