
第12讲 The Immune System Gone Wrong
HEADS UP!
Most of the time, the immune system functions flawlessly, but occasionally it "makes mistakes." In some situations, the immune system may mobilize weapons which are not appropriate for the situation. And in rare instances, the immune system may mistake friend for foe, and attack our own bodies.
INTRODUCTION
Thus far, we have focused on the good that the immune system does in protecting us from infection. Occasionally, however, the immune system "goes wrong" – sometimes with devastating consequences. In this lecture we will examine several situations in which the immune system plays a major role in producing the damaging effects (the pathology) of a disease.
DISEASES CAUSED BY DEFECTS IN IMMUNE REGULATION
Roughly a quarter of the U.S. population suffers from allergies to common environmental antigens (allergens) that either are inhaled or ingested. Hay fever and asthma are the two most common allergic diseases of the respiratory tract. Hay fever is caused by proteins that are derived from mold spores or plant pollens. These allergens are present in the outside air, usually at certain times of the year. In contrast, the allergens that cause asthma
are mostly found indoors. Dust mites, cockroaches, rodents, and household pets are major sources of these allergy-causing proteins. In addition to allergies caused by allergens in the air we breathe, the food we eat also can cause allergies.
The immune systems of non-allergic people respond weakly to these allergens, and produce mainly antibodies of the IgG class. In striking contrast, allergic individuals (called atopic individuals ) produce large quantities of IgE antibodies. Indeed, the concentration of IgE antibodies in the blood of those with allergies can be 1,000- to 10,000-fold higher than that in the blood of non-atopic people!
It is the overproduction of IgE antibodies in response to otherwise innocuous environmental antigens that causes allergies.
In Lecture 3, we discussed the interaction of IgE antibodies with white blood cells called mast cells.
Because mast cell degranulation is a central event in many allergic reactions, let's take a moment to review this concept. When atopic individuals first are exposed to an allergen (e.g., pollen) they produce large amounts of IgE antibodies which recognize that allergen. Mast cells have receptors on their surface that can bind to the Fc region of IgE antibodies. Consequently, after the initial exposure, mast cells will have large numbers of these allergen-specific IgE molecules attached to their surface. Allergens are small proteins with a repeating structure to which many IgE antibodies can bind close together. So on a second or subsequent exposure, an allergen can crosslink the IgE molecules that are bound to the mast cell surface, dragging the mast cell's receptors together. This clustering of IgE receptors tells mast cells to degranulate – to release their granules, which normally are stored safely inside the mast cells, into the tissues in which they reside. Mast cell granules contain histamine and other powerful chemicals and enzymes that can cause the symptoms with which atopic individuals are intimately familiar.
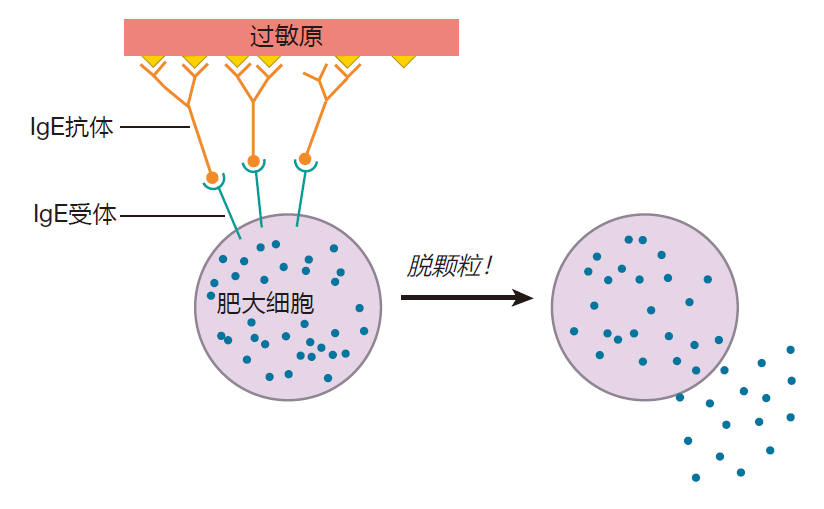
Interestingly, although IgE antibodies have a half-life of only about two days in the blood, once they are attached to mast cells, they have a half-life of weeks to months.
This means that mast cells can stay "armed" and ready to degranulate for an extended period after exposure to an allergen.
Allergic reactions generally have two phases: immediate and delayed. The immediate reaction to an allergen is the work of mast cells which are stationed out in the tissues and basophils, another granule-containing white blood cell, which can be recruited from the blood by signals given off by mast cells responding to an allergen.
Like mast cells, basophils have receptors for IgE antibodies, and crosslinking of these receptors can lead to basophil degranulation.
Although mast cells and basophils are responsible for the immediate reaction to an allergen, a third granulecontaining white blood cell, the eosinophil, is the prominent player in chronic allergic reactions (e.g., in asthma).
Before an "attack" by an allergen, there are relatively few eosinophils present in the tissues or circulating in the blood. However, once an allergic reaction has begun, helper T cells secrete cytokines such as IL-5, which can recruit many more eosinophils from the bone marrow.
These eosinophils can then add their "weight" to the allergic reaction. Because eosinophils must be mobilized from the marrow, their contribution is delayed relative to that of mast cells and basophils, which can respond almost immediately.
Of course, mast cells and basophils did not evolve just to annoy atopic people. These cells, with their ability to degranulate "on command," provide a defense against parasites (e.g., worms) that are too large to be phagocytosed by professional phagocytes. In a sense, IgE antibodies act as a "guidance system" for these cells, targeting their weapons to the enemy. For example, by discharging their destructive chemicals directly onto the skin (tegument) of a parasite to which IgE antibodies have bound, mast cells can destroy these massive creatures.
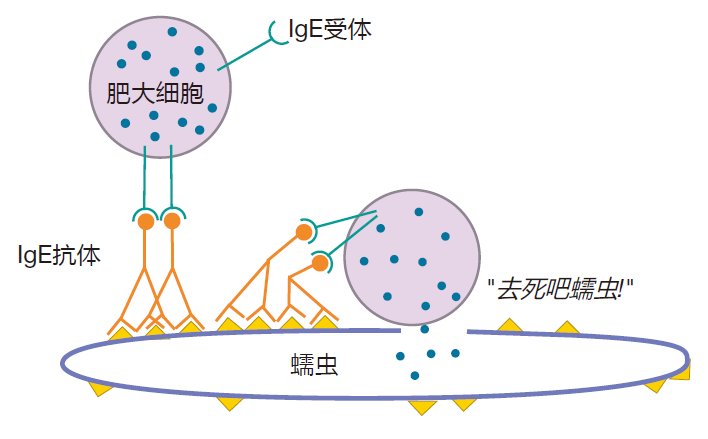
What makes this defense so elegant is that in response to a parasitic infection, parasite-specific IgE antibodies are made, and mast cells and basophils are armed. However, nothing happens unless these armed cells come in contact with a parasite that can cluster their IgE receptors. Consequently, you don't get uncontrolled degranulation, wreaking havoc throughout your body. Rather, the IgE guidance system allows these cells to zero in on parasites, causing relatively little collateral damage to our tissues.
Why do some people have allergies?
It is clear that IgE antibodies are the bad guys in allergic reactions, but what determines whether a person will make IgE or IgG antibodies in response to an allergen?
You remember from Lecture 6 that helper T cells can be "instructed" by the environment in which they are stimulated to secrete various cytokine subsets (e.g., Th1, Th2, or Th17). And the cytokines given off by these T cells can then influence B cells undergoing class switching to produce IgA, IgG, or IgE antibodies. For example, a germinal center that is populated with Th1 cells usually will produce B cells that make IgG antibodies, because Th1 cells secrete IFN-γ, which drives the IgG class switch.
In contrast, B cells tend to change to IgE production if class switching takes place in germinal centers that contain Th2 cells which secrete IL-4 and IL-5. Consequently, the decision to produce either IgG or IgE antibodies in response to an allergen will depend heavily on the type of helper T cells present in the secondary lymphoid organ which intercepts the allergen. Indeed, helper T cells from allergic individuals show a much stronger bias toward the Th2 type than do Th cells from non-atopic people.
The hygiene hypothesis
So atopic individuals produce IgE antibodies because their allergen-specific helper T cells tend to be of the Th2 type. But how do they get that way? The answer to this important question is not known for certain, but many immunologists believe that a bias toward Th2-type helper T cells can be established early in childhood, and in some cases, even before birth. Here's how this is thought to work.
A fetus inherits roughly half of its genetic material from its mother and half from its father. As a result, the fetus is really a "transplant" that expresses many paternal antigens to which the mother's immune system is not tolerant. Since the placenta is the interface between the mother and the fetus, measures must be taken to avoid having maternal CTLs and NK cells attack the placenta because it expresses these paternal antigens. The Th1 subset of helper cells secretes TNF, which helps activate NK cells, and IL-2, which causes NK cells and CTLs to proliferate. So it would be advantageous for the survival of the fetus to bias maternal Th cells away from the Th1 cytokine profile. Indeed, cells of the placenta produce relatively large amounts of IL-4, which influences maternal helper T cells to become Th2 cells. Importantly, these same placental cytokines also have a strong influence on fetal helper T cells. As a result, most humans are born with helper T cells that are strongly biased toward making Th2 cytokines.
Obviously this bias does not last a lifetime, and eventually most people end up with a more balanced population of Th1 and Th2 cells. One event that probably helps establish this balance is infection at an early age with microbes (e.g., viruses or bacteria) that normally elicit a Th1 response. Indeed, it is suspected that early microbial infections may be important in "reprogramming" a child's immune system so that a Th1 response to allergens results. Immunologists hypothesize that if a microbial infection strongly biases the immune response toward a Th1 type at the same time that the child encounters an allergen (say, a dust mite protein), the Th response to that allergen also will be deviated toward the Th1 type. Once this deviation takes place, feedback mechanisms tend to lock in the Th1 bias, and memory T cells will be generated that remember not only the allergen, but also their Th1 response to it. Once a large number of biased memory cells is built up, it is difficult to reverse this bias, so early exposure to infectious diseases may be critical in establishing a normal reaction to environmental allergens. Said another way, the early-life "education" of the immune system may have a large impact on the health of an individual later in life.
The idea that childhood microbial infections or early exposure to allergens might be important in biasing our immune systems toward producing Th1 helper T cells in response to environmental allergens is called the hygiene hypothesis. Indeed, in Western countries, where improved personal hygiene has led to a decrease in child-hood infections, and where exposure to certain allergens early in life is less frequent, the incidence of allergies to environmental allergens has increased dramatically.
Some of the best data in support of the hygiene hypothesis comes from studies of families living on traditional farms. These studies have shown that children who have contact with farm animals have a significantly lower incidence of asthma and hay fever than do children from rural areas who do not live on a farm. This effect is even more pronounced if their mothers come in contact with multiple animal species and animal feed (such as hay and grain) during their pregnancy. Interestingly, the timing of exposure to animals and their feed seems to be important, with the greatest protection being observed for children who live on a farm during the first few years of life. In this regard, it is important to note that living on a farm is not "unusual" as far as the human immune system is concerned. After all, until recently, many of our ancestors lived in close contact with farm animals, and it is likely that the immune system evolved to function best – at least in terms of allergies – in that setting.
Although immunologists call it the "hygiene hypothesis," it actually should be called the "lifestyle hypothesis" –because the increased incidence of allergies is likely due to a change in lifestyle, not just to improved hygiene. For example, prior to the 1950s, when televisions became commonplace in the United States, most children came home from school and went outdoors to play. Things are very different now, with many young children spending long hours indoors playing video games or staring at a television. Also, after World War II, it became customary for childhood illnesses to be treated with antibiotics, which can destroy intestinal bacteria that are helpful in setting up a "balanced" immune system. Indeed, studies show an increased risk of asthma in children who were treated with multiple courses of antibiotics during their first year of life.
It also has been proposed that regulatory T cells may help bias the immune system away from the production of IgE antibodies in response to environmental allergens.
Helper T cells out in the tissues can be induced to become regulatory T cells (iTregs). Consequently, one could argue that if a person is routinely exposed to environmental allergens, some of his CD4 + T cells may be induced to become regulatory T cells, which could suppress the immune response to these allergens. Indeed, inducible regulatory T cells produce IL-10 and TGFβ – cytokines which are known to bias antibody production away from IgE and toward IgG or IgA. Moreover, in individuals who are not atopic, regulatory T cells represent the majority of CD4 + T cells that are specific for common environmental allergens.
Heredity
In addition to environmental factors (e.g., early exposure to infectious diseases or environmental allergens), heredity clearly plays a large part in susceptibility to allergies.
For example, if one identical twin suffers from allergies, the probability is about 50% that the second twin also will be atopic. Immunologists have noticed that people who are allergic to some allergens are more likely to have inherited certain versions of the class II MHC genes than are non-atopic people, suggesting that these particular MHC molecules may be especially efficient at presenting these allergens. Also, some atopic individuals produce mutant forms of the IgE receptor. It is hypothesized that these mutant receptors may send an unusually strong signal when crosslinked, resulting in secretion of abnormally high levels of IL-4 by mast cells – which favor the production of IgE antibodies. Unfortunately, mutations in genes that confer susceptibility to allergies have been difficult to identify – because there seem to be many of them, and because they usually differ from atopic individual to atopic individual.
The best current synthesis of this information is that the immunological basis for allergies is a defect in immune regulation in which allergen-specific helper T cells are strongly polarized toward a Th2 cytokine profile, resulting in the production of allergen-specific IgE antibodies. The genes a person inherits can make him more or less susceptible to allergies, and exposure to environmental factors such as microbial infections may influence whether susceptible individuals become atopic.
It is important to recognize that allergies were not a major issue for our ancestors. They were not blessed with the good hygiene we have today, and were exposed at an early age to infectious diseases that most likely deviated their response to allergens away from the abnormal production of IgE antibodies. In fact, while many Americans may curse IgE antibodies, people in much of the rest of the world depend heavily on these antibodies to defend them against parasites. Parasitic worms still infect roughly a third of the human population.
Treatments for allergies
Although not a cure, treatment with glucocorticoid steroids can decrease allergy symptoms by blocking cytokine production by helper T cells. As a result, fewer B cells are activated (because they do not get the help they need), and the total number of antibodies made is reduced. Steroids, however, are not specific for allergies, and steroid treatment decreases the number of activated B cells of all kinds. Consequently, taking glucocorticoid steroids for extended periods can result in increased susceptibility to infectious diseases. Recently, immunologists have produced antibodies (e.g., omalizumab) which can grasp the Fc region of IgE antibodies and block the binding of these antibodies to mast cells. Treatment with these antibodies is quite effective in relieving allergic symptoms and decreasing the severity of asthma attacks.
So far, only one approach – specific immunotherapy –has been successful in curing allergies. This treatment involves the injection of gradually increasing doses of crude extracts of allergens until a maintenance dose is achieved. After several years of regular injections, some patients become tolerant to the allergen (or allergens) in the extract. The immediate result of these injections is that mast cells become more difficult to activate in response to IgE binding. Then, over time, these injections encourage allergen-specific B cells to switch their antibody class from IgE to one of the other antibody classes. Indeed, during specific immunotherapy, the ratio of IgG to IgE antibodies specific for the allergen being administered can increase 10- to 100-fold. Unfortunately, the mechanisms by which this immune deviation is achieved are not well understood. The latest thinking is that repeated injections of allergen extracts may generate inducible regulatory T cells which produce cytokines that suppress IgE antibody production. This view is supported by the finding that beekeepers, who receive repetitive doses of bee venom (because they are stung frequently), do not suffer severe allergic reactions when stung by bees, and have elevated levels of IL-10 – a cytokine produced by iTregs.
AUTOIMMUNE DISEASE
The human immune system does not expend a huge amount of biological "energy" on a foolproof system in which every B and T cell is carefully checked for tolerance of self. Instead, the system relies on a multilayered strategy in which each layer includes mechanisms that should weed out most self-reactive cells, with lower layers catching cells that slip through tolerance induction in the layers above. This usually works very well, but occasionally "mistakes are made," and instead of defending us against foreign invaders, the weapons of our immune system are turned back on us. Autoimmune disease results when a breakdown in the mechanisms meant to preserve tolerance of self is severe enough to cause a pathological condition. Roughly 5% of Americans suffer from some form of autoimmune disease.
Some cases of autoimmunity result from genetic defects.
For example, most autoimmune diseases are chronic disorders that involve repeated stimulation of self-reactive lymphocytes. In healthy people, this is controlled by activation-induced cell death in which chronically stimulated T cells are eliminated when Fas proteins on their surface are ligated. Humans with genetic defects in either the Fas or the Fas ligand protein lack this layer of tolerance protection, and their T cells refuse to die when chronically stimulated by self antigens. The resulting disease, autoimmune lymphoproliferative syndrome or Canale–Smith syndrome, has, as its pathological consequences, massive swelling of lymph nodes, the production of antibodies that recognize self antigens, and the accumulation of a large number of T cells in the secondary lymphoid organs.
Although some autoimmune disorders are caused by genetic defects, the majority of autoimmune diseases occur when the layers of tolerance-inducing mechanisms fail to eliminate self-reactive cells in genetically normal individuals. In fact, you could argue that the potential for autoimmune disease is the price we must pay for having B and T cell receptors which are so diverse that they can recognize essentially any invader.
The latest thinking is that for autoimmunity to occur, at least three conditions must be met. First, an individual must express MHC molecules that efficiently present a peptide derived from the target self antigen. This means that the MHC molecules you inherit can play a major role in determining your susceptibility to autoimmune disease. For example, only about 0.2% of the U.S. population suffers from juvenile diabetes, yet for Caucasian Americans who inherit two particular versions of class II MHC genes, the probability of contracting this autoimmune disease is increased about 20-fold.
The second requirement for autoimmunity is that the affected person must produce T and, in some cases, B cells which have receptors that recognize a self antigen.
Because TCRs and BCRs are made by a mix-and-match strategy, the repertoire of receptors that one individual expresses will be different from that of every other human, and will change with time as lymphocytes die and are replaced. Even the collections of TCRs and BCRs expressed by identical twins will be different. Therefore, it is largely by chance that a person will produce lymphocytes whose receptors recognize a particular self antigen.
So for autoimmune disease to occur, a person must have MHC molecules that can present a self antigen, and lymphocytes with receptors that can recognize the self antigen – but this is not enough. There also must be environmental factors that lead to the breakdown of the tolerance mechanisms which are designed to eliminate self-reactive lymphocytes. For years, physicians have noticed that autoimmune diseases frequently follow bacterial or viral infections, and immunologists believe that microbial attack may be one of the key environmental factors that triggers autoimmune disease. Now clearly, a viral or bacterial infection cannot be the whole story, because for most people, these infections do not result in autoimmunity. However, in conjunction with a genetic predisposition (e.g., the type of MHC molecules inherited) and lymphocytes with potentially self-reactive receptors, a microbial infection may be the "last straw" that leads to autoimmune disease.
Molecular mimicry
Immunologists' current favorite hypothesis to explain why infections might lead to the breakdown of self tolerance is called molecular mimicry. Here's how this is thought to work.
Lymphocytes have BCRs or TCRs that recognize their cognate antigen. It turns out, however, that this is almost never a single antigen. Just as one MHC molecule can present a large number of peptides which have the same overall characteristics (length, binding motif, etc.), a TCR or a BCR usually can recognize (cross react with) several different antigens. Generally, a TCR or BCR will have a high affinity for one or a few of these cognate antigens, and lower affinities for the others.
During a microbial invasion, lymphocytes whose receptors recognize microbial antigens will be activated.
The molecular mimicry hypothesis holds that sometimes these receptors also recognize a self antigen, and if they do, an autoimmune response to that self antigen may result. It is presumed that before the microbial infection, these potentially self-reactive lymphocytes had not been activated – either because the affinity of their receptors for the self antigen was too low to trigger activation, or because the restricted traffic patterns of virgin lymphocytes never brought them into contact with the self antigen under conditions that would promote activation.
Self-reactive B cells could also be generated by molecular mimicry during somatic hypermutation. This could happen if the receptors of a B cell that originally recognized only a bona fide pathogen mutated so that they could recognize both the pathogen, making them "eligible" for Tfh help, and a self antigen, making them potentially destructive.
In these scenarios, the invading microbe substitutes for (mimics) the self antigen for activation. And once activated in response to a cross-reacting microbial antigen, these self-reactive lymphocytes can do real damage.
Cross-reactive antibodies have been identified in some patients with autoimmune disease who previously had been infected by certain viruses or bacteria. For example, it is believed that rheumatic heart disease, which is a possible complication of a streptococcal throat infection, can result when receptors on helper T cells that recognize streptococcal antigens cross react with a protein which is present on the tissues that make up the mitral valve of the heart. These cross-reactive Th cells may then direct an inflammatory response that can severely damage this heart valve.
One reason it has been so difficult to pin down the environmental triggers for most autoimmune diseases is that TCRs which recognize self antigens usually can cross react with multiple environmental antigens. Consequently, although viral or bacterial infections may be involved in some autoimmune disorders, it appears unlikely that any single microbe is responsible for any one autoimmune disease.
Animal models of human autoimmune diseases have been useful for understanding which immune system players are involved, which self antigens are targets of the immune response, and which microbial antigens might be involved in the molecular mimicry that may trigger disease. Typically, these models involve animals that have been bred to be exquisitely susceptible to autoimmune disease, or animals whose genes have been altered to make them susceptible. Nevertheless, animal models frequently differ in important respects from the human disease they are meant to model. As a result, many treatments for autoimmune diseases which looked promising in an animal model have turned out to be useless in humans.
Inflammation and autoimmune disease
Although molecular mimicry may result in the activation of lymphocytes that previously had been ignorant of self antigens, these self-reactive lymphocytes still face a problem once they reach the tissues where the self antigen is located: They must be reactivated before they can do any real damage. If the innate immune system is battling an infection in the tissues, inflammatory cytokines such as IFN-γ and TNF secreted by cells of the innate system will activate APCs (e.g., macrophages) that reside in the tissues. Once activated, these APCs express the MHC and co-stimulatory molecules required to re-stimulate T cells which enter the tissues to do battle. Consequently, when lymphocytes venture out into the tissues to join a war that the innate system is already fighting, re-stimulation is not a problem. However, for a T cell that recognizes a self antigen which the innate system does not see as dangerous, the tissues can be a very inhospitable place – because that self-reactive lymphocyte usually will not receive the co-stimulation necessary for its survival.
What this means is that it is not enough for a microbe to activate self-reactive T cells by mimicry. There also must be an inflammatory reaction going on in the same tissues that express the self antigen. Otherwise it is unlikely that self-reactive lymphocytes would exit the blood into these tissues, or that they would survive if they did. This requirement for inflammation at the site of an autoimmune attack helps explains why, for example, a strep infection in the throat only rarely leads to rheumatic heart disease.
So one scenario that immunologists favor for the initiation of autoimmune disease is this: A genetically susceptible individual is attacked by a microbe that activates T cells whose receptors just happen to cross react with a self antigen. Simultaneously, an inflammatory reaction takes place in the tissues where the self antigen is expressed.
This inflammation could be caused either by the mimicking microbe itself, or by another, unrelated infection or trauma. As a result of this inflammatory reaction, APCs are activated that can re-stimulate self-reactive T cells.
In addition, cytokines generated by the inflammatory response can upregulate class I MHC expression on normal cells in the tissues, making these cells even better targets for destruction by self-reactive CTLs.
Examples of autoimmune disease
Autoimmune diseases are usually divided into two groups: organ-specific and systemic diseases. Let's look at examples of both types, paying special attention to the self antigens against which the autoimmune response is thought to be directed, and to the environmental antigens that may be involved in molecular mimicry.
Insulin-dependent diabetes mellitus (type 1 or "juvenile" diabetes) is an example of an organ-specific autoimmune disease. In this disease, the targets of autoimmune attack are the insulin-producing β cells of the pancreas.
Although antibodies produced by self-reactive B cells may participate in the chronic inflammation that contributes to the pathology of this disease, it is currently believed that the initial attack on the β cells is mediated by CTLs.
In diabetes, destruction of β cells usually begins months or even years before the first symptoms of diabetes appear, so this disease is sometimes referred to as a "silent killer." Indeed, by the time of diagnosis, more than 90% of a patient's β cells usually will have been destroyed. Until insulin injections became possible in the 1920s, the life expectancy of someone diagnosed with diabetes was a matter of months. Even now, with the use of supplementary insulin, the disease shortens average life expectancy by more than a decade.
Antibodies that bind to β cell antigens are produced very early in the disease. As a result, relatives of diabetic patients can be tested to determine whether they might be in the initial stages of diabetes, and could be helped by early intervention. Indeed, if a child has a sibling who developed diabetes early in life, and if that child's immune system does make antibodies which recognize beta cell proteins, the probability that he will develop diabetes within the next five years is nearly 100%.
Clearly, there are genetic factors that help determine susceptibility to diabetes, since the probability that both identical twins will suffer from this autoimmune disease is about 50% if one of them has it. It is known, for example, that some individuals have a version of the gene for CTLA-4 which is associated with an increased risk of type 1 diabetes. Patients with this variant make less CTLA-4 RNA, and presumably are less able to limit the activation of self-reactive T cells that recognize β cell antigens.
Thus far, no strong candidates have emerged for environmental factors that might trigger the initial attack on β cells. However, many immunologists believe that diabetes results, at least in part, when the balance between natural regulatory T cells and potentially self-reactive CTLs is upset. Indeed, mutations in genes that compromise nTreg function can cause autoimmune disease both in humans and in mice.
Plaque psoriasis is an autoimmune disease that affects about 2% of the U.S. population. The most noticeable symptoms are the thickening and scaling of the surface of the skin. In the most severe cases, these "plaques" can cover more than 10% of the skin area. Recently, it was discovered that this disease is driven by CD8 + T cells that produce high levels of IL-17 (yes, killer T cells can produce cytokines!). IL-17 can bind to skin cells (keratinocytes) and set off a chain of molecular events which causes keratinocytes to proliferate to form plaques. A current "molecular mimicry" model is that T cells in genetically susceptible individuals (e.g., those who have certain versions of the class I MHC molecules) have receptors that recognize both a particular keratin protein and a protein made by streptococcal bacteria (which infect a large proportion of the world's population from time to time).
It is hypothesized that some CTLs responding to such a streptococcal infection have TCRs that cross react with the keratin protein presented by the patient's class I MHC molecules. Once activated by the bacterial infection, these self-reactive CD8 + T cells produce IL-17, causing errant keratinocyte proliferation and plaque formation. These particular versions of class I MHC molecules may have been selected for during evolution because they help the immune system protect against streptococcal infections. After all, for a caveman, a streptococcal infection could be deadly, yet cavemen probably didn't care much about the appearance of their skin!
Rheumatoid arthritis is a systemic autoimmune disease that affects approximately 1% of the world's population. It is characterized by chronic inflammation of the joints. One of the presumed targets of this autoimmune reaction is a certain cartilage protein, and T cells from arthritic patients can recognize both the cartilage protein and a protein encoded by the bacterium that causes tuberculosis.
IgM antibodies that can bind to the Fc region of IgG antibodies are abundant in the joints of individuals with rheumatoid arthritis. These antibodies can form IgM–IgG antibody complexes, which can activate macrophages that have entered the joints, increasing the inflammatory reaction. Indeed, the inflammation associated with rheumatoid arthritis is caused mainly by tumor necrosis factor produced by macrophages which infiltrate the joints under the direction of self-reactive helper T cells. Interestingly, mice injected with Mycobacterium tuberculosis develop inflammation of the joints, suggesting, but not proving, that a TB infection may trigger rheumatoid arthritis in some patients.
Celiac disease or gluten intolerance is an organ-specific autoimmune disease that affects about 1% of the people in Europe and the United States. Gluten or gluten-like proteins are found in wheat, barley, and rye. The normal immune response in the small intestine to these innocuous food antigens is the activation of gluten-specific inducible regulatory T cells (iTregs) that enforce tolerance of these antigens. In contrast, the intestinal immune system of individuals who are gluten intolerant mounts a strong inflammatory response which damages the villi of the intestinal epithelium. These villi are responsible for the absorption of nutrients, and the damage results in diarrhea and unintended weight loss. Essentially everyone with celiac disease has inherited one or both of two particular versions of the class II MHC molecules, and these two types of MHC molecules have been shown to efficiently present gluten peptides to helper T cells. Nevertheless, only about 4% of people who have these particular MHC molecules have celiac disease. So owning these MHC molecules is necessary but not sufficient for gluten intolerance.
Celiac disease is interesting in that it is not caused by molecular mimicry. Although recent experiments suggest that celiac disease may be "triggered" when genetically susceptible individuals are infected by common intestinal viruses (e.g., reovirus), the antigen that helper T cells recognize is not an antigen that is "shared" by gluten and a virus. Rather, the disease results when the intestinal immune system somehow becomes "confused." Instead of viewing gluten as an innocuous food protein, and producing gluten-specific regulatory T cells, the immune system sees gluten as a dangerous invader, and mobilizes a Th1 inflammatory response. Exactly how this happens remains a mystery.
Finally, lupus erythematosus is a systemic autoimmune disease that affects about 250,000 people in the United States, roughly 90% of whom are women. This disease can have multiple manifestations including a red rash on the forehead and cheeks (giving the "red wolf" appearance for which the disease was named), inflammation of the lungs, arthritis, kidney damage, hair loss, paralysis, and convulsions. Lupus is caused by a breakdown in both B and T cell tolerance that results in the production of a diverse collection of IgG antibodies which recognize a wide range of self antigens, including DNA, DNA–protein complexes, and RNA–protein complexes.
These autoantibodies can form self antigen–antibody complexes which "clog" organs in the body that contain "filters" (e.g., kidneys, joints, and the brain), causing chronic inflammation.
Non-identical twins have about a 2% probability of both having lupus if one twin has the disease. With identical twins, the probability is increased about ten-fold. This indicates a strong genetic component to the disease, and more than a dozen MHC and non-MHC genes have been identified – each of which seems to slightly increase the probability that a person will contract lupus. Although no specific microbial infection has been associated with the initiation of this autoimmune disease, mice that lack functional genes for Fas or Fas ligand exhibit lupus-like symptoms. This has led immunologists to speculate that lupus may involve a defect in activation-induced cell death, in which lymphocytes that should die due to chronic stimulation survive to cause the disease. There is also some evidence that humans with mutations which increase the sensitivity of their Toll-like receptors to RNA or DNA are lupus-prone. The idea here is that recognition of human DNA by a B cell's receptors, together with an unusually strong signal from a mutated Toll-like receptor, could be misinterpreted as a dangerous situation. As a result, B cells could be activated without T cell help, and anti-DNA antibodies could be produced.
REVIEW
Sometimes the immune response may be misguided.
Indeed, allergies result when the immune system produces IgE antibodies – which are designed to deal with a parasitic infection – in response to environmental antigens. Immunologists are not sure what causes this misguided response. Their best thought is that a defect in immune regulation causes production of a large number of allergen-specific Th2 cells. These helper T cells then orchestrate the overproduction of allergen-specific IgE antibodies. Atopic individuals frequently inherit a "genetic landscape" which predisposes them to allergies, and the timing and extent of exposure to pathogens may influence whether susceptible individuals become atopic.
In fact, the hygiene hypothesis holds that if the immune system of a young child is not appropriately challenged by microbial infections, allergies may result.
Autoimmunity occurs when the mechanisms designed to enforce tolerance of self antigens don't function properly. In some cases, this is the result of genetic defects.
However, in most cases, immunologists don't know what causes the breakdown in tolerance-inducing mechanisms.
Clearly, for autoimmunity to occur, a person must have MHC molecules which can present self antigens, and lymphocytes with receptors that can recognize these antigens.
So there is a genetic component. In addition, it is believed that environmental factors are involved, although such factors have been difficult to discover – probably because there are so many of them. It is hypothesized that autoimmunity can be triggered when an invading microbe "mimics" a self antigen. According to this scenario, the microbe activates lymphocytes which have receptors that recognize both a microbial antigen and a self antigen. Once activated in response to the microbial invasion, these cross-reactive lymphocytes can lead an attack on both the invader and the cells or proteins belonging to the infected individual.
Inflammation also is believed to play a role in molecular mimicry by providing the signals required to attract cross-reactive lymphocytes and keep them activated.