
4.5 细胞衰老的代谢基础
你已经了解到衰老的原因与物理学的基本定律有关,细胞紊乱随时间的增加会导致受损蛋白质的积累。受损蛋白质的积累,即分子去保真度,导致细胞功能下降,最终导致细胞自我毁灭,即细胞衰老(随着时间的推移,细胞凋亡在细胞死亡中的作用似乎越来越小)。我们现在将注意力转向可能导致分子去保真度的损害的衰老机制。在本节中,我们将介绍细胞最基本的功能(即能量代谢)如何产生可能造成损害的副产物。
当大气中的氧气含量增加时,多细胞生物就会出现
大约在25亿到30亿年前,蓝绿藻(蓝藻)的细胞开始利用太阳光的能量将大气中的二氧化碳(CO2)转化为葡萄糖。在这个被称为光合作用的过程中,氧气(O2)被释放到大气中。随着氧气开始累积,两个关键事件发生,使得生命出现了多样性。首先,太阳的辐射能将O2转化为臭氧(O3),臭氧在高层大气中积累并捕获有害的紫外线。生命现在能够离开海洋的保护,在陆地上定居。第二,由于基于氧的细胞呼吸,即有氧呼吸,比无氧呼吸效率更高,因此使用有氧呼吸的单细胞生物体变得更大,并更成功地竞争可用资源。这些单细胞有氧呼吸生物比无氧呼吸细胞上的成功使得多细胞生物的进化。
不幸的是,对许多生物来说,大气中氧气的积累是致命的。O2的分子结构使其对不能安全地将有氧代谢的副产物还原为水的生物体具有极高的毒性。没有抗氧化保护的生物体很快就会死亡,只剩下那些能够保护自己免受氧气有害影响的生物体。换句话说,适应性最高的生物体是那些可以利用氧气为其带来好处的生物体。
1956年,Denham Harman提出理论,随着细胞衰老,有氧代谢的一些副产物被称为氧中心自由基,氧代谢的产物具有一个或多个未配对电子,它们逃逸了正常的降解途径,并对生物分子造成损害。反过来,这些受损分子在细胞中积累,导致细胞衰老。他的理论,氧化应激理论,曾经被认为是衰老的主要机制。我们现在知道,氧化应激只是导致细胞损伤进而导致年龄相关功能障碍的众多机制之一。由于有大量关于氧应激引起的损伤的信息,我们仅将这一过程作为衰老机制的一个例子,衰老导致细胞损伤的积累和细胞衰老的诱导。这一过程不应被视为导致细胞损失从而导致细胞衰老的主要或唯一原因。我们在本章末尾提出的案例表明,涉及活性氧物种(ROS)的进化保守机制是细胞抵御应激的方式之一。我们认为氧化应激是拮抗性多效性的一个例子。
氧化代谢产生活性氧
在解释氧中心自由基的性质之前,简要回顾一下将有机燃料转化为细胞能量的氧化还原反应可能会有所帮助。电子从一种化合物流向下一种化合物,构成了我们如何从营养物质(脂肪、碳水化合物和蛋白质)中产生可用细胞能量的基础,并决定了化合物的反应性。物质失去一个或多个电子时被氧化;当它获得一个或多个电子时,它就会被还原。在任何涉及两种物质之间电子流动的反应中,氧化和还原必须协同工作。当一种物质失去一个电子(氧化),另一种物质获得一个电子(还原)。氧化化合物往往具有高度的反应性,因为它们从其他化合物中寻找电子以稳定其电子构型。还原化合物往往比氧化化合物更稳定,因为它们的电子构型更接近基态。
生物体中的氧化—还原过程可被描述为氧和氢的丢失或获得。如果一种化合物获得氧气或失去氢气,它就被氧化了。如果它失去氧气或获得氢气,它就会被还原。在生物化学中,化合物的氧化-还原通常与第三个分子有关,这一过程称为耦合氧化-还原反应。图4.16显示了丙酮酸和乳酸的耦合氧化-还原。通过烟酰胺腺嘌呤二核苷酸(NAD+)还原为NADH+H+,乳酸被氧化(失去氢)形成丙酮酸。相反,当NADH+H+氧化为NAD+时,丙酮酸还原为乳酸。
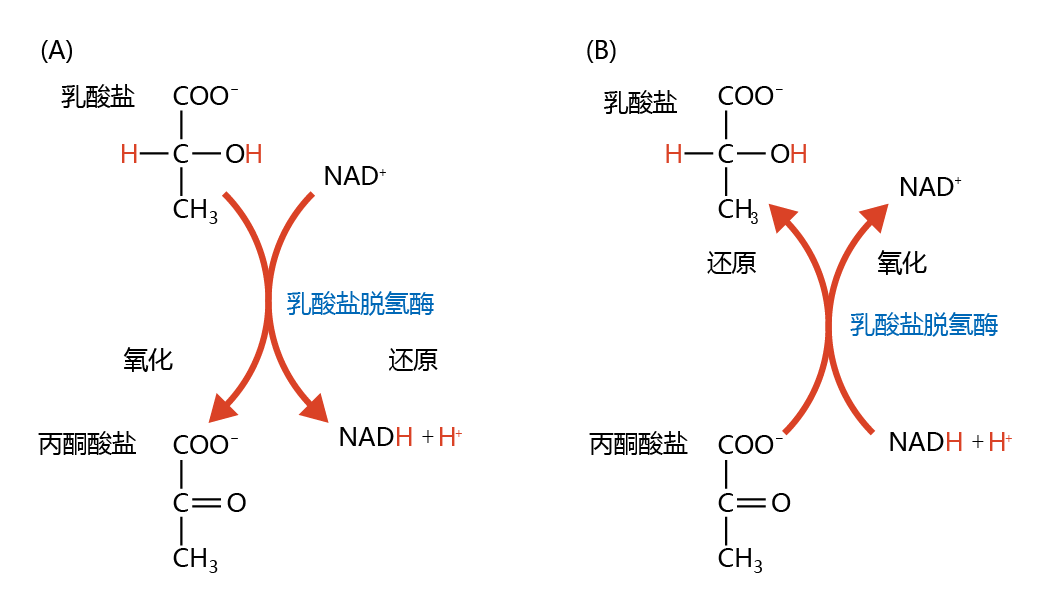
图4.16 丙酮酸和乳酸的耦合氧化-还原反应。(A)在乳酸氧化为丙酮酸的过程中,乳酸的两个氢(红色)被烟酰胺腺嘌呤二核苷酸(NAD+)的氧化形式除去,形成还原形式NADH+H+。(B)相反,在NADH+H+氧化为NAD+的反应中加入两个氢,丙酮酸就会还原为乳酸。
这篇关于氧化还原反应的简短描述也可能帮助你理解抗氧化剂是如何工作的,这是自由基化学中的一个重要概念。回想一下,氧化化合物比还原化合物具有更少的电子,使它们更具反应性。抗氧化剂是一种化合物,它向被氧化的分子提供电子,使其更易还原,活性降低。在本节中,我们将讨论过氧化氢酶、维生素E和维生素C等抗氧化剂如何保护细胞免受氧化化合物和自由基的伤害。
氧中心自由基,也称为活性氧(ROS),包括超氧自由基(•O2−), 过氧化氢(H2O2)和羟基自由基(•OH)。虽然双原子氧(O2)的基态被归类为自由基,但它的反应性很弱。氧在不同的轨道上有两个未配对的电子,它们可以匹配的自旋。因此,O2只能与具有两个自旋与未配对氧电子自旋反平行的未配对电子的其他原子发生反应,这在自然界中很少见。因此,氧的还原是有氧代谢的一个重要过程,每次必须有一个电子发生反应(must occur one electron at a time)。氧的单电子还原会产生一个高活性分子,它有一个未配对的电子(记得你之前读到的关于电子损失和活性的内容),称之为超氧自由基(•O2−); •O2−的一个额外的单电子还原会产生同样具有破坏性的过氧化氢(H2O2)分子。有氧呼吸生物体已经发展出酶机制,允许氧气完全还原为无害化合物,如H2O(图4.17)。活性氧及其还原可发生在细胞的不同部位,包括线粒体、细胞核和胞浆。 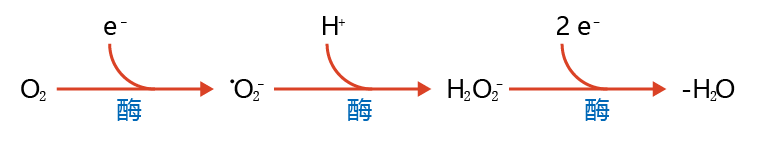
图4.17 酶将氧气(O2)还原为水(H2O)的过程。在正常有氧代谢过程中,氧被还原(加一个电子)形成超氧自由基(•O2−)。酶几乎在瞬间将•O2−催化变成过氧化氢(H2O2)。反过来,其他酶催化H2O2的双电子还原生成H2O。
线粒体ATP的合成过程产生大部分超氧离子
有氧生物的线粒体(图4.18)利用身体消耗的95%的氧气来合成ATP。如图4.19所示,该分子是为细胞反应提供化学能的主要分子。以最简单的形式,氧化代谢将能源物质(糖和脂肪)的碳-碳键中储存的能量转化为可用能量,即ATP,蛋白质在有氧生物体中很少用作能量。ATP转化为ADP释放出能量驱动许多生化反应。
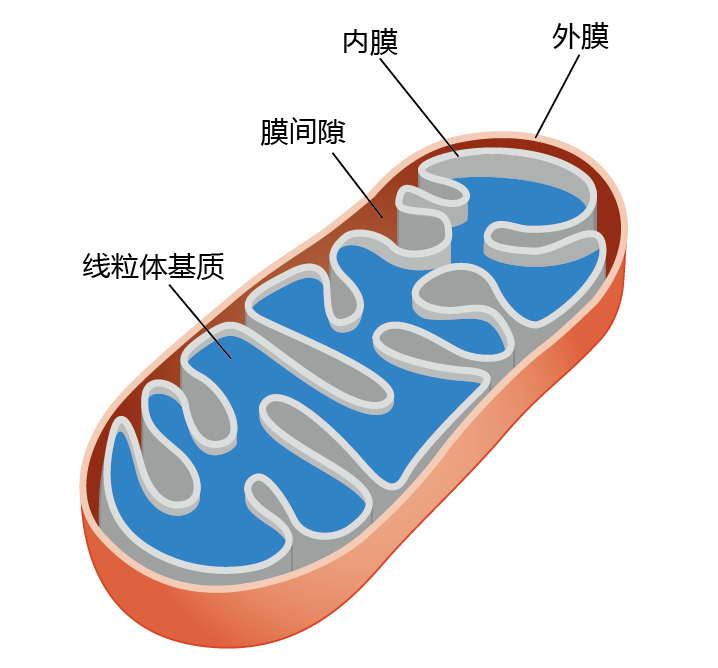
图4.18 线粒体。外膜将线粒体与细胞质分离,并含有与脂肪代谢有关的酶。为了增加与电子传递和ATP合成有关的反应的表面积,内膜被折叠。基质中含有用于氧化磷酸化的酶和线粒体DNA。膜间空间含有ATP从线粒体中运出线粒体所需的酶。
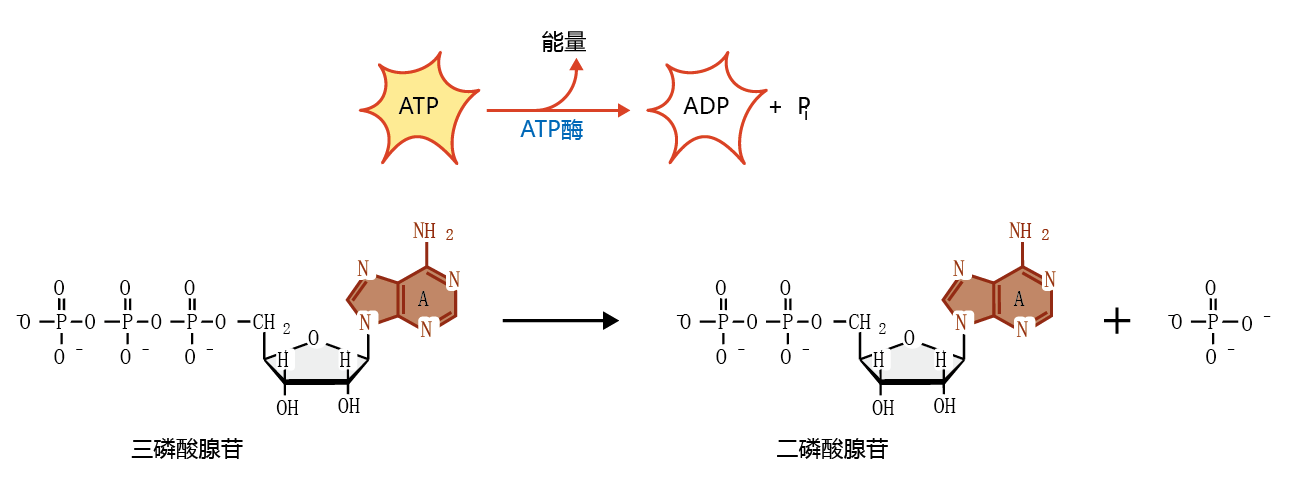
图4.19 三磷酸腺苷(ATP)和二磷酸腺苷(ADP)的结构。在ATP转化为ADP的过程中,一个高能磷酸键的断裂释放出的能量为细胞代谢提供了大部分化学能。
能量从营养物质转移到ATP发生在线粒体内两个不同的但耦合的系统中。第一个系统通过一系列氧化还原反应生成电子,这是三羧酸循环(TCA循环)的一部分,也被称为Krebs循环和柠檬酸循环。TCA循环发生于线粒体的基质中。TCA循环通过一系列酶催化的氧化作用从碳-碳键释放电子。然后这些电子被“穿梭”到线粒体内膜上的第二个系统,即电子转移系统(ETS)。ETS利用这些电子,通过一系列酶催化还原,为ATP合成提供必要的能量(图4.20)。整个过程称为氧化磷酸化。
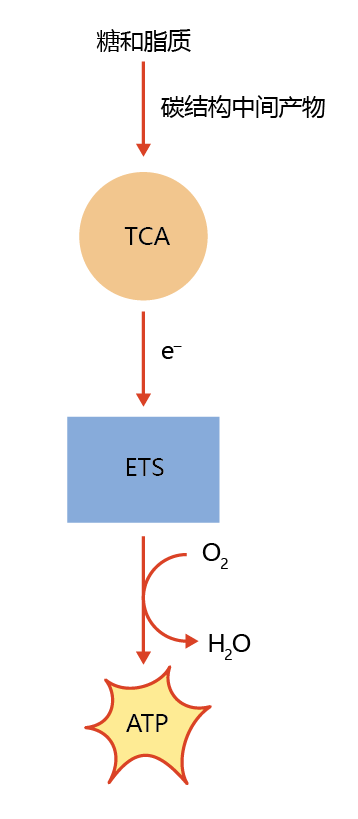
图4.20 食物能量(碳水化合物和脂质)产生的细胞能量(ATP)。碳水化合物和脂质中的碳-碳键的能量不能被细胞直接利用。通过三羧酸(TCA)循环中含碳中间体的还原,电子被释放,并在电子转移系统(ETS)中与氧反应,合成三磷酸腺苷(ATP)。
如图4.21所示,当来自营养物质碳水化合物或脂肪的碳被用于生成乙酰辅酶A分子时,氧化磷酸化开始。在TCA循环的特定点,被称为电子(能量)载体或还原等效物的化合物[如烟酰胺腺嘌呤二核苷酸(NAD+)和黄素腺嘌呤二核苷酸(FAD)]“拾取”氧化反应中释放的电子。这些电子传递链将电子传递给ETS。
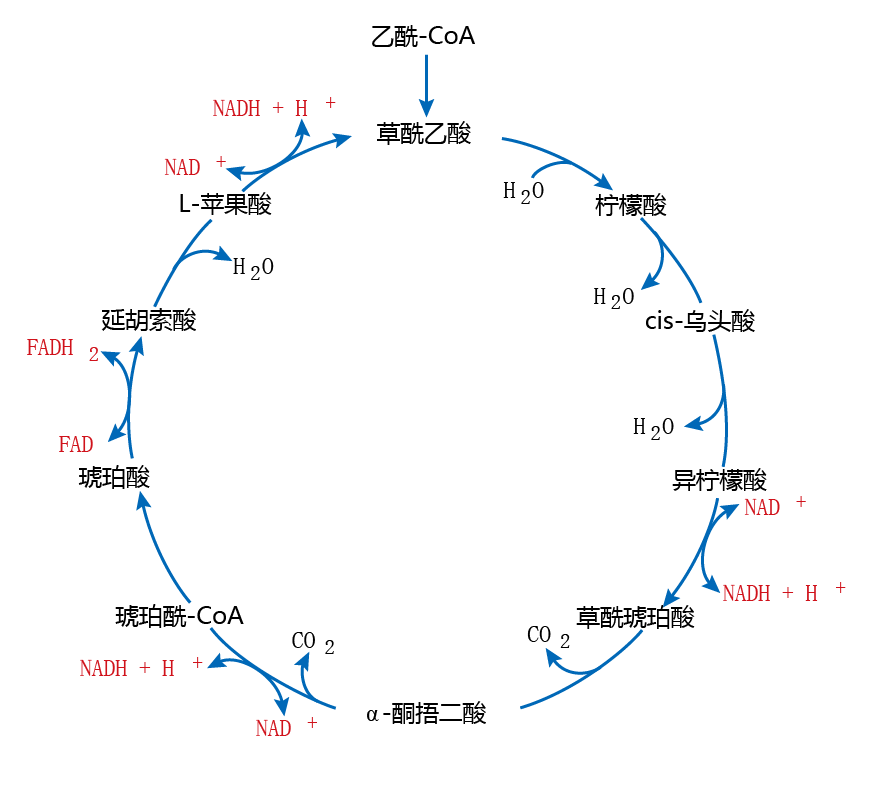
图4.21 三羧酸(TCA)循环。食物中的碳水化合物和脂肪被氧化成乙酰辅酶A,然后进入TCA循环。在一系列氧化-还原反应中,电子生成之后并由电子载体NADH+H+和FADH2(此处显示为红色)传递到电子传递系统(见图4.19)。
在理解ETS如何帮助ATP的合成时,需要从物理学角度思考并记住三个要点:(1)与电子相关的自由能驱动系统,(2)必须在线粒体基质和膜间隙之间建立质子(H+)梯度,(3)膜间隙中的质子浓度必须大于基质中的质子浓度。TCA循环中产生的电子通过还原等价物NADH+H+和FADH2输送至ETS,并通过氧化反应释放(图4.22)。这些质子被特殊的蛋白质通过内膜“泵送”到膜间隙。因为线粒体内膜不允许质子渗透,所以必须依靠这些特殊蛋白质来跨膜运输。内膜的不渗透性是建立质子梯度所必需的。电子提供了驱动质子泵运行的自由能。当电子放弃自由能时,质子梯度变得更大。随着电子在ETS内转移,维持梯度的自由能降低,质子流回基体。但这种流动只能发生在膜上的特点部位,即ATP合成酶,它能催化生成ATP的反应。
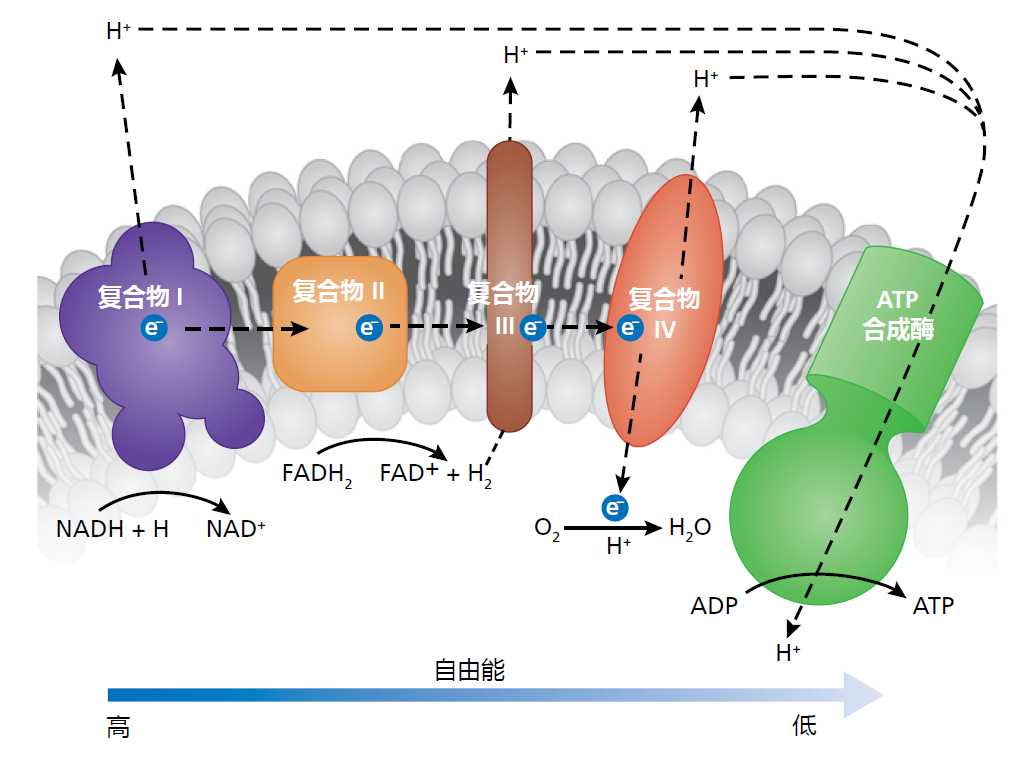
图4.22 电子转移系统(ETS)和ATP合成的简化版本。高能的电子驱动三个呼吸复合体,这些呼吸复合体将质子泵入膜间隙并建立质子梯度。在这个过程中,自由能减少,质子(H+)通过ATP合成酶流回来,因此驱动ATP合成酶用来合成ATP。氧气与质子生成水,并且ATP合成后留下的自由能散失。
任何系统的能量状态都会努力达到平衡。因此,即使线粒体基质的低自由能状态也必须散失。在此处,氧气有它的作用。氧以电子和质子的形式作为自由能的最终受体,最后产生水。虽然这个系统在将氧气完全还原为水方面非常有效,但有一些氧仅仅被一个电子还原,然后会生成一个超氧自由基。
在ATP合成过程中,氧气作为电子的最终受体,这个过程效率非常高,它是靠TCA循环与ETS的合作完成的。也就是说,当TCA循环活跃时,很少(如果有的话)产生超氧化物自由基。然而,当线粒体基质中ATP/ADP比率较高时,ATP的合成速率就会降低,此时ETS中的条件有利于产生超氧化物自由基•O2− (图4.23)。线粒体基质中的较高ATP/ADP比率抑制了TCA循环,电子不会穿梭到ETS。质子没有流过ATP合成酶,因此氧气没有被还原成水。基质中的低浓度质子抑制了氧气还原成水的过程。因此,氧可以被一个电子还原成•O2−,这个电子由富含电子(还原)的ETS复合物提供。 估计表明,在细胞消耗的氧气中,大约0.5%–1%的氧气会产生超氧化物自由基。
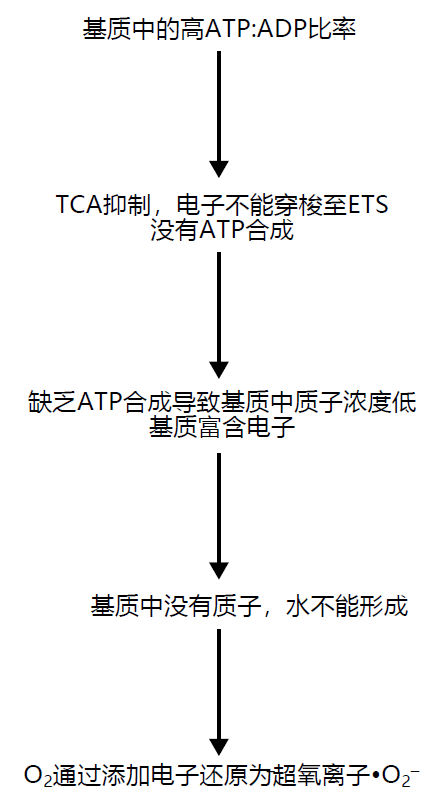
图4.23 线粒体中生成超氧自由基•O2−的步骤。
酶将超氧化物自由基还原为水
如前所述,•O2-可在正常有氧代谢过程中生成。如果未能及时清除,这些自由基会与其他原子迅速反应,导致细胞受损的可能性。幸运的是,对于需氧生物来说,线粒体含有两种酶,可以将•O2-还原为水,这两种酶是超氧化物歧化酶(SOD)和过氧化氢酶。超氧化物歧化酶对•O2-有很高的亲和力, 使得该自由基立即还原为H2O2(图4.24)。然而,H2O2也会对细胞造成严重伤害。此时还原级联中的第二种酶,也就是过氧化氢酶,对H2O2进行双电子还原,并且形成H2O。
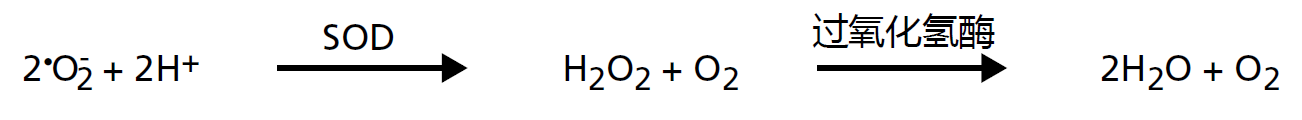
图4.24 线粒体中超氧化物自由基还原成水的过程。超氧化物歧化酶(SOD)将两个超氧化物自由基还原为过氧化氢(H2O2)。然后过氧化氢酶将H2O2还原为水。
细胞质基质中的还原也会产生自由基
虽然细胞质基质中的氧浓度远低于线粒体,但单电子还原仍能产生超氧离子。发生这种情况时,细胞质基质中的•O2− 被超氧化物歧化酶和过氧化氢酶还原为水,这一反应与线粒体中发生的反应类似(如图4.24所示)。细胞质基质中的H2O2也可以在谷胱甘肽过氧化物酶的帮助下还原为水(图4.25)。然而,在某些细胞质基质条件下,过氧化氢转化为高度活性的羟基自由基•OH。当细胞质基质中同时存在铁或铜(Fe2+或Cu2+)和H2O2时,这种转化以非酶方式发生,这种反应称为Fenton反应(图4.26A);当Fe2+、H2O2和•O2− 在细胞质基质中同时存在,这一过程称为Haber-Weiss反应(图4.26B)。
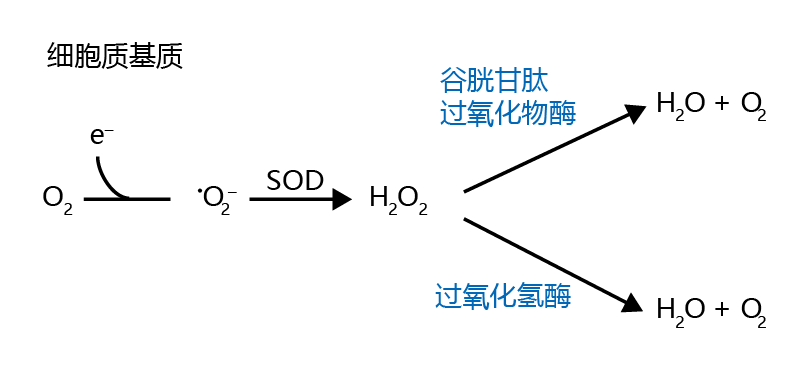
图4.25 在细胞质基质中超氧化物离子还原为水。氧气还原为•O2− 随后通过细胞质基质SOD将超氧化物还原为H2O2。过氧化氢随后通过过氧化氢酶或谷胱甘肽过氧化物酶还原为H2O和O2。
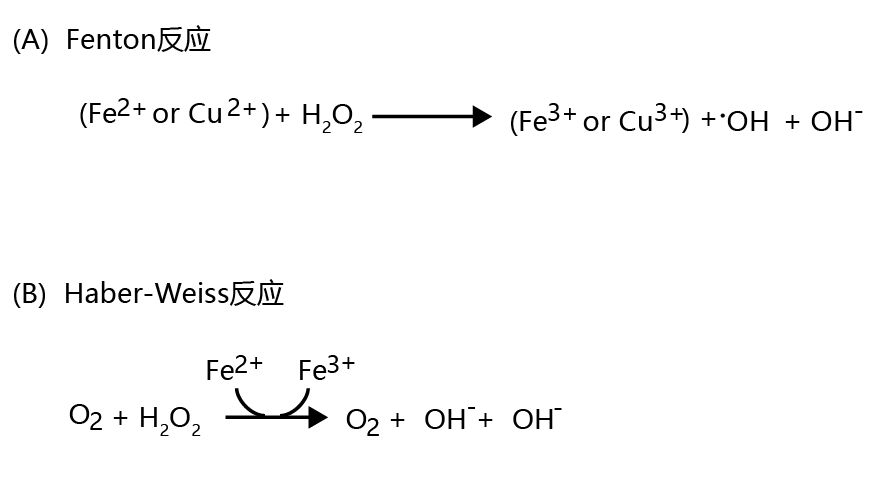
图4.26 细胞质基质中羟基自由基(•OH)的生成。在(A)Fenton反应和(B)Haber-Weiss反应中,过氧化氢可转化为高活性羟基自由基•OH。这两个反应都以非酶方式进行。
氧中心自由基(Oxygen-centered free radicals)导致受损生物分子的积累
自由基非常活跃,会导致许多生物分子的结构发生重大变化,如核酸、脂质和蛋白质。反过来,结构的改变导致分子的生物活性降低。这种损伤可以在细胞组织的各个层次上观察到(图4.27)。脂质过氧化物在膜磷脂中的积累使得细胞膜和线粒体膜在维持细胞外和细胞内隔室之间的屏障方面效果变差。反过来,对溶质和水的浓度敏感的化学反应也会受到影响。DNA转录和翻译过程中的组分氧化损伤导致蛋白质中氨基酸序列改变。此外,由于ROS还可以影响DNA修复机制的蛋白质,不适当的碱基对取代可能无法被移除,从而无法匹配正确的碱基。似乎参与从细胞中去除受损蛋白质的蛋白质也受到ROS的影响。也就是说,细胞清除废物功能的下降加剧了受损蛋白质的积累。
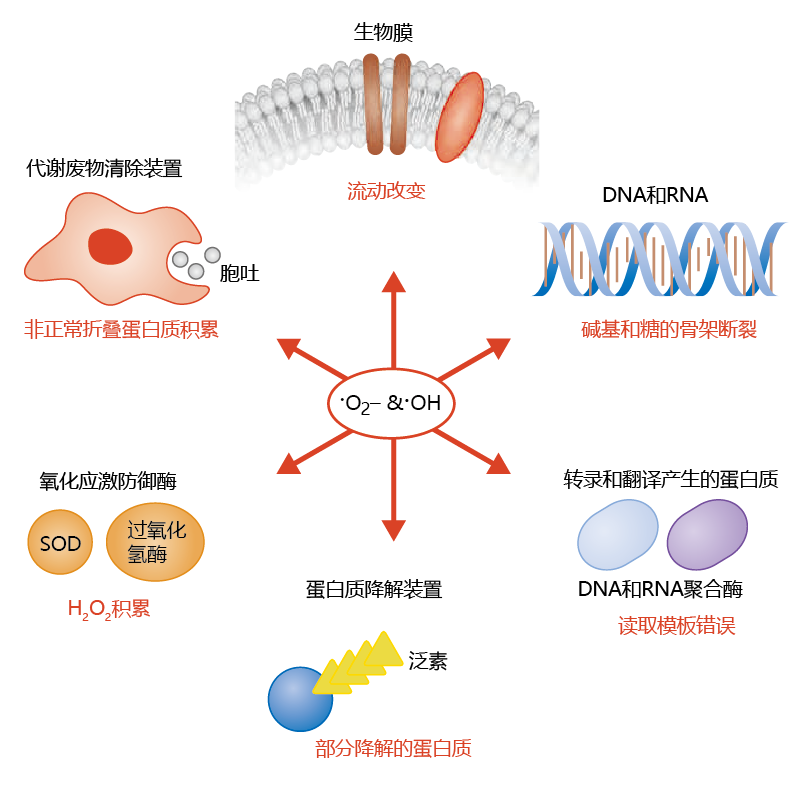
图4.27 活泼的氧自由基可以导致破坏的生物分子在几乎所有的细胞组织中积累。
尽管线粒体产生的大部分细胞ROS是•O2−, 但是线粒体的抗氧化系统(SOD和过氧化氢酶)的效率使得极少数的超氧化物离子引起损伤。相反,细胞质的活性氧(H2O2和•OH)似乎才是自由基造成的大多数细胞损伤的原因,尽管它们仅占细胞总活性氧的15%。这说明:•OH与多不饱和脂肪反应非常迅速(在1×10−12秒内),多不饱和脂肪是一种主要存在于生物膜中的化合物,但在细胞其它位置也有(见本章后面的讨论)。
以氧为中心的自由基也可以导致DNA碱基对序列的改变。虽然大多数研究发现,在衰老细胞中,由于自由基损伤而导致的DNA错误是罕见的,但羟基自由基(•OH)确实对DNA的糖骨架和鸟嘌呤之间的键有很高的亲和力(图4.28)。例如,如果错误发生在复制过程中,腺嘌呤可以代替鸟嘌呤。
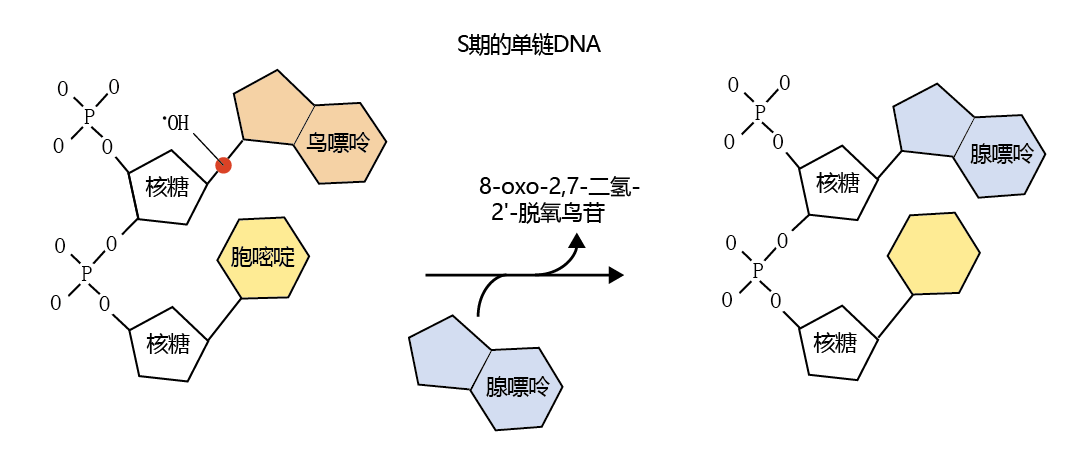
图4.28 羟基自由基(•OH)对DNA的影响。羟基自由基,如图中的红点所示,可以在复制过程中破坏鸟嘌呤和DNA糖骨架之间的键。如果DNA的修复机制不能识别这个突变,复制的DNA就会改变序列。碱基对断裂的副产物:8-氧代-2,7-二氢-2′-脱氧鸟苷(8-oxo-2,7-dihydro-2′-deoxyguanosine)可作为DNA损伤的量度。
在DNA复制过程中,腺嘌呤取代鸟嘌呤将导致G-C对被替换成A-T对。随之蛋白质的氨基酸序列也可能会发生改变,这可能导致生物活性降低或受损蛋白质的积累。
细胞膜容易受到活性氧的破坏
细胞膜在细胞内和细胞外隔室之间保持高效的屏障,同时允许细胞功能所必需的分子交换。细胞膜的这种特性是因为脂肪酸(脂类)的化学结构和物理排列以及膜中嵌入的各种分子形成的。膜脂质被称为磷脂,因为它们含有一个水溶性磷酸盐头部和两个水不溶性脂肪酸尾部(图4.29)。细胞膜也称为脂质双层,因为磷脂排列在双层中,使得一层磷脂的磷酸端指向细胞外空间,而另一层的磷酸端则指向细胞内空间。磷脂的脂质成分在膜的内部相互指向。这种排布确保水和水溶性分子不会在细胞外和细胞内空间之间自由通过,这是维持细胞适当化学平衡的重要特性。水溶性分子通过嵌入在膜中的转运蛋白进入或离开细胞。
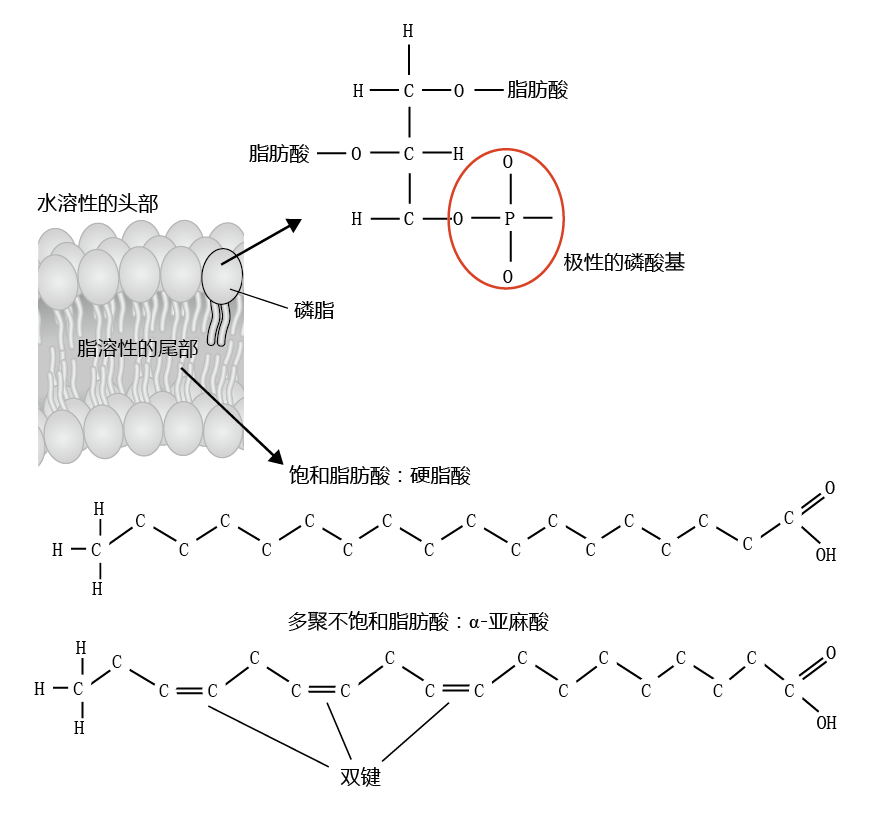
图4.29 细胞膜中磷脂的结构。磷脂头部的极性磷酸基团是水溶性或亲水性的。脂肪酸尾部不溶于水或疏水。磷脂可以含有两种饱和脂肪酸、两种不饱和脂肪酸或每种脂肪酸中的一种。生物膜中的大多数磷脂都只有一种:要么是饱和脂肪酸,要么是不饱和脂肪酸。
功能复杂的膜在很大程度上取决于磷脂的物理排列和膜中的结构。如果膜结构和磷脂之间的粘附太紧密,水不溶性分子就不能自由通过膜。如果粘附太松散,那么不适当数量的水和水溶性分子会进入细胞或从细胞中逸出。因此,双层膜已经进化,使得脂质和蛋白质处于完美的平衡,从而为正确的膜结构实现适当的电化学成分。这种平衡的任何变化都会改变膜功能,从而改变细胞功能。ROS引起的生物膜变化可导致膜渗透性的破坏、电解质梯度的不平衡、正常膜蛋白迁移率的抑制以及对正常细胞活动至关重要的各种其他功能的破坏。
细胞膜的流体性质在很大程度上反映了磷脂中所含脂肪酸的单双键特性。磷脂是饱和脂肪酸和不饱和脂肪酸的混合物,它们结合在一起使膜具有特定的粘度。饱和脂肪酸在体温下比不饱和脂肪酸具有更大的粘度。因此,将两者结合成磷脂可赋予膜流动性,从而实现最佳功能。
细胞膜脂质成分中的多不饱和脂肪的双键结构特别容易受到•OH的“攻击”。•OH对多不饱和脂肪的“攻击”会引发链式反应,传播额外的自由基,并将多不饱和脂改造成新的分子,即脂质过氧化物。如图4.30所示,•OH将其未配对的电子提供给多不饱和脂肪(LH)的双键,形成脂质自由基(L•)和水。该反应标志着自由基链式反应的起始阶段。L•可以减少胞质的O2,并产生脂质过氧化物自由基(LOO•),LOO•进而攻击另一个LH,导致产生L•和脂质过氧化物(LOOH),并进一步生成LOO•、LOOH和L•。
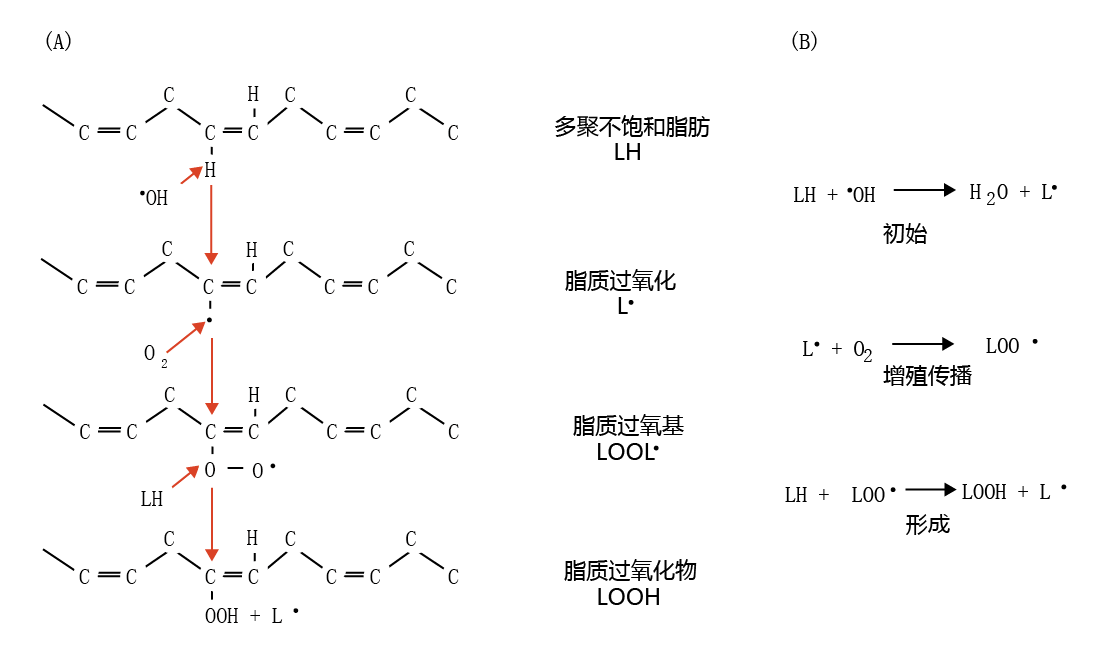
图4.30 脂质过氧化物的形成。(A)羟基自由基“攻击”多不饱和脂肪(LH)的双键,从而形成脂质自由基(L•)。反过来,胞质O2与脂质自由基的未配对电子反应,形成脂质过氧化物自由基(LOO•)。脂质过氧化物自由基攻击另一种多不饱和脂肪,形成脂质过氧化物分子(LOOH)和新的脂质自由基,等等。(B)自由基链式反应的引发和传播导致脂质过氧化物的形成。
如果产生脂质过氧化物的脂质自由基反应不受调控,细胞功能将受到极大的阻碍,甚至完全停止。幸运的是,细胞已经发展出一种机制来防止脂质过氧化物的形成,其中包括维生素E(生育酚)和维生素C(抗坏血酸)。维生素E存在于细胞膜附近或细胞膜内,对羟基自由基和脂质自由基的亲和力远大于膜多不饱和脂肪的双键亲和力。如图4.31所示,通过将α-维生素E还原为α-维生素E自由基,脂质过氧化物自由基被氧化为LOOH,从而终止脂质过氧化物链式反应。然而,α-维生素E自由基需要被氧化回还原的α-维生素E。这是通过多步骤过程实现的,需要维生素C(抗坏血酸)、谷胱甘肽和NAD+的参与。
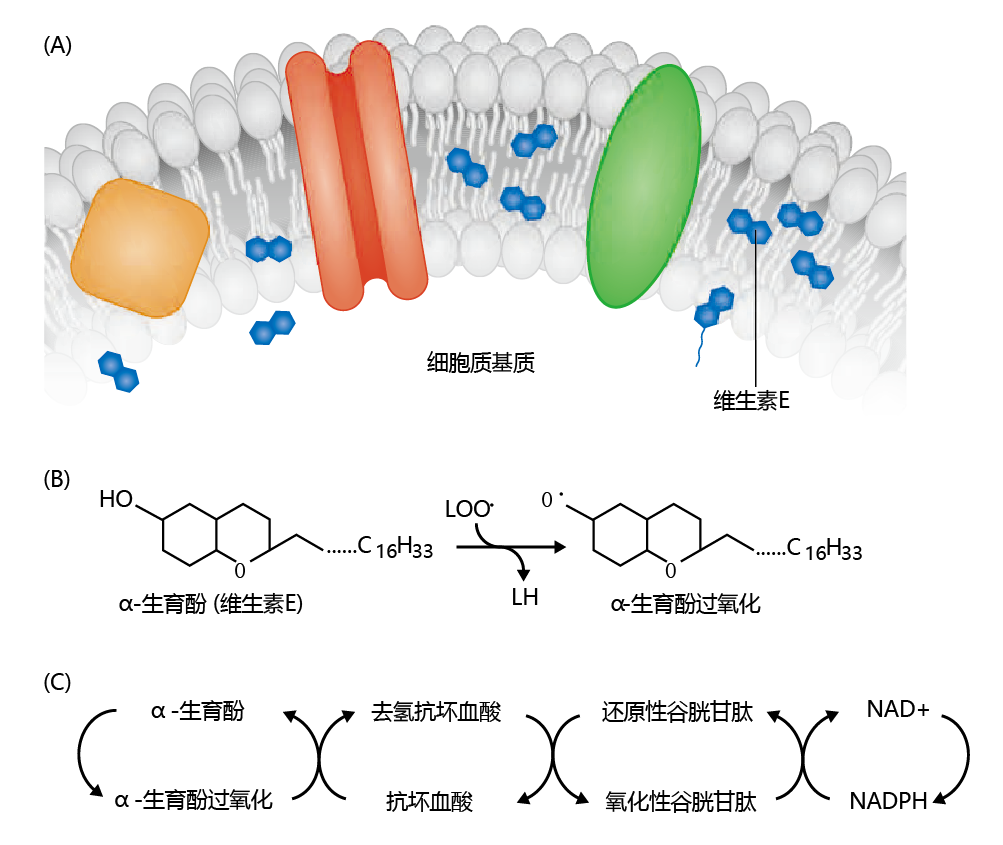
图4.31 维生素E(α-生育酚)终止自由基链式反应。(A)维生素E位于细胞膜内或附近。(B)如果出现自由基,维生素E在脂质过氧化物自由基氧化过程中被还原为α-生育酚自由基。(C)α-生育酚在抗坏血酸、谷胱甘肽和NAD+的参与下,通过多个步骤被再次氧化。
拮抗多效性解释了导致活性氧物种损伤的衰老机制
到目前为止,我们的讨论对ROS描绘了一幅相当黯淡的画面。这种消极的局面很可能是由于过去50年来人们对ROS的破坏性进行了大量研究。从哈曼的衰老氧化理论开始,许多生物学家就质疑ROS损伤作为衰老机制的进化基础。也就是说,自然选择不会保留那些即使对细胞造成最轻微的损害的基因,因为那样就不会有生殖优势了。因此,ROS必须具有有益的目的,为发育中的有机体提供益处。如果ROS对机体有好处,并且它们确实有好处(如下文所述),那么在衰老细胞中观察到的ROS损伤就可以通过拮抗多效性来解释了(见第3章)。
回想一下,G·C·威廉姆斯的拮抗性多效性理论预测,在生命早期表达有益健康的基因将被选择,即使它们在晚年可能是不利的。ROS有几种有益的作用,可以向生物体传递繁殖优势。例如,免疫系统使用ROS作为破坏外来有害物质的机制。当外来入侵者(如细菌)进入血液或组织时,免疫系统会从淋巴结中释放大量细胞,攻击并摧毁入侵者。其中一种细胞类型是巨噬细胞。巨噬细胞释放酶产生的分子,这些分子旨在破坏入侵生物体的细胞结构。这些分子中包括•O2−, 其主要工作似乎是通过产生脂质过氧化物部分的破坏侵略者的细胞膜(图4.32,顶部)。最近的研究还表明,ROS和/或ROS还原的副产物,如H2O2,可通过诱导抗氧化剂:超氧化物歧化酶和过氧化氢酶的进一步表达来防止氧化损伤(图4.32,底部)。尽管确切的机制尚不清楚,但在体外培养细胞中,如果诱导它们产生超氧自由基的话,它们似乎激活了SOD和过氧化氢酶基因的启动子区。与巨噬细胞功能或SOD和过氧化氢酶表达相关的蛋白质分子保真度的随机损失可能导致细胞和组织•O2− 平衡和氧化损伤。也就是说,尽管在以后的生活中可能会有一些损害,即拮抗性多效性,但生物进化仍然选择了赋予ROS产生益处的基因。
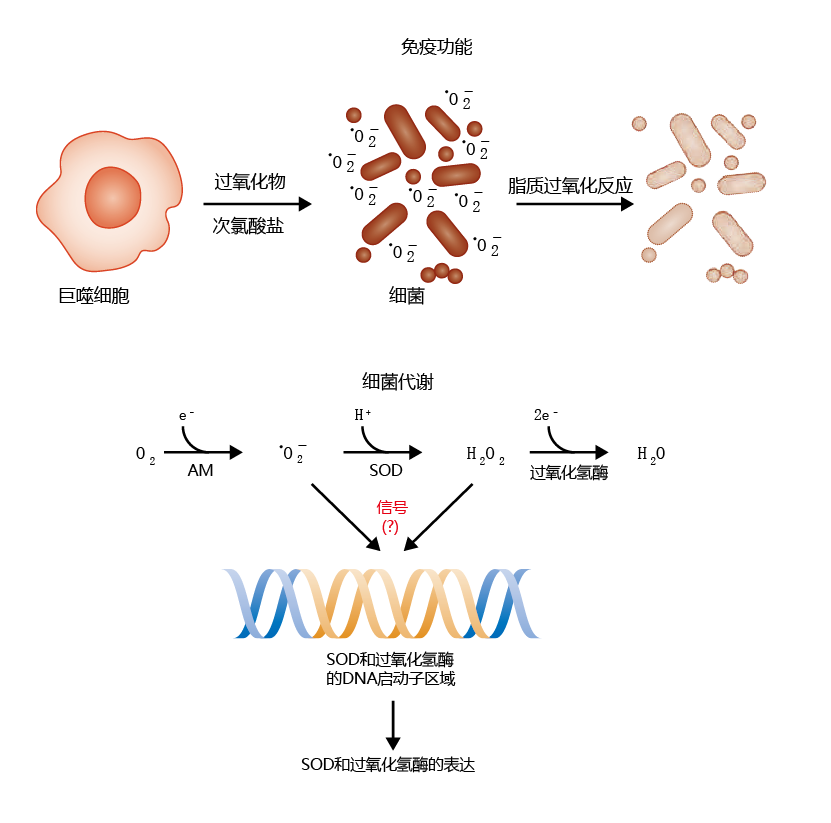
图4.32 ROS对生物体有益的两个例子。免疫功能(上图):免疫系统中的巨噬细胞分泌有毒化学物质,如超氧自由基和次氯酸钠,以应对入侵的生物。超氧化物离子有助于通过脂质过氧化途径破坏细菌的细胞膜。有氧代谢(底部):正常有氧代谢过程中ROS的产生也可以刺激超氧化物歧化酶(SOD)和过氧化氢酶的表达,尽管确切的机制尚不清楚。
ROS损伤反映了为生殖优势而选择的蛋白质分子保真度的随机损失,这一假设得到了相当大的支持,因为发现ROS介导低氧反应,这是细胞对低氧水平的保护机制。低氧反应由低氧诱导因子1α(HIF-1α)调节(图4.33)。在正常的氧水平下,在氧化磷酸化增加的过程中,在几乎没有任何ROS产生的情况下,HIF-1α在其达到完全功能状态之前被细胞内的酶降解。(蛋白质降解机制在第5章中讨论。)缺氧或低水平氧化磷酸化过程中胞浆中ROS的存在抑制HIF-1α的降解。然后,一个具有完整功能的HIF-1α进入细胞核,在那里它刺激与增加氧摄取和保护细胞免受氧化损伤有关的几种蛋白质的表达。有600多种蛋白质参与维持细胞内正常的氧水平,毫无疑问,ROS在正常细胞功能中起着重要作用。我们在第5章中表明,HIF-1α途径的改变可以对寿命产生重大影响,这种影响与ROS损伤无关。
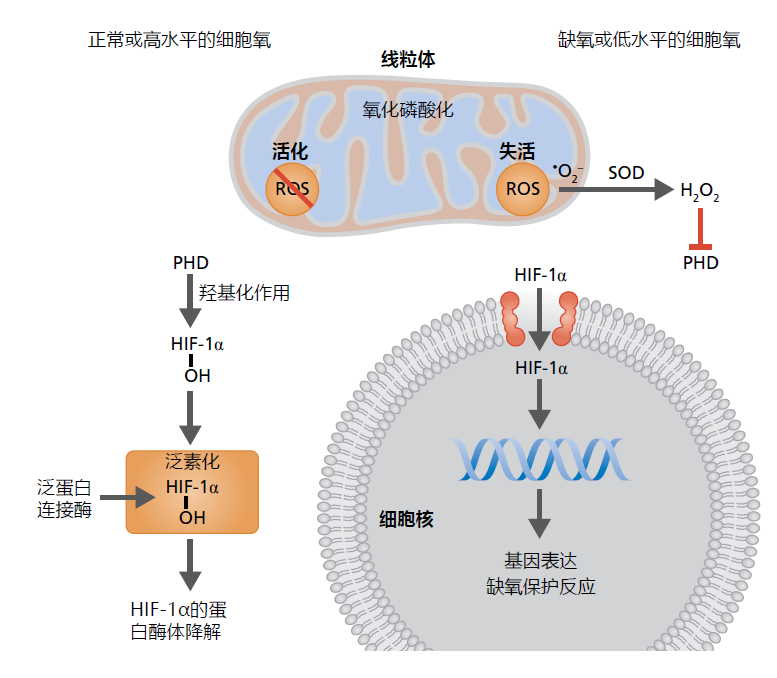
图4.33 活性氧物种在低氧反应中的作用。当细胞中的氧水平处于正常浓度或增加(左)时,例如在运动期间,HIF-1α被脯氨酸羟化酶结构域蛋白(PHD)羟基化。HIF-1α的羟基化状态作为泛素蛋白酶体蛋白降解途径的标志(见第5章),HIF-1β在完全发挥功能之前被降解。缺氧或低水平的细胞氧降低氧化磷酸化,从而增加•O2− 并通过SOD还原为H2O2;线粒体膜对H2O2是可渗透的。H2O2一旦进入胞浆,就会抑制PHD的作用,从而使HIF-1α成为一种完全功能的蛋白质。HIF-1α进入细胞核,在那里与DNA上的位点结合,这些结合位点会引起好几种低氧保护蛋白的表达。