
8.2 CHANGES IN SKELETAL MUSCLE
There are 640 skeletal muscles in the human body, comprising 40%–50% of the total body weight. Skeletal muscle's primary purpose involves movement and stability. Thus, time-dependent alterations in skeletal muscle mass, function, or both, can have a significant impact on one's dexterity, locomotion, and balance. In this section we explore the possible changes to adult skeletal muscle over time. We begin by describing the organization of skeletal muscle and the mechanism of contraction. Then, we discuss time-dependent alterations in skeletal muscle, including the effects of exercise on aging muscle. Finally, we briefly describe sarcopenia, an age-related pathological process involving excess loss in skeletal muscle mass.
Muscle contraction is result of molecular interactions between actin and myosin proteins within sarcomere
Skeletal muscles have several levels of organization, and each level may be affected by time (Figure 8.8). Our discussion here focuses primarily at the microscopic level where the interaction of the contractile proteins, actin and myosin, causes the muscle to shorten (contract). Single actin molecules are coiled long, thin, flexible polymers of the globular actin protein (Figure 8.9A). The globular portions of the actin molecule align in the same direction, giving the filament a negative charge at one end and a positive charge at the other. This polar arrangement of the actin filament is critical to contraction.
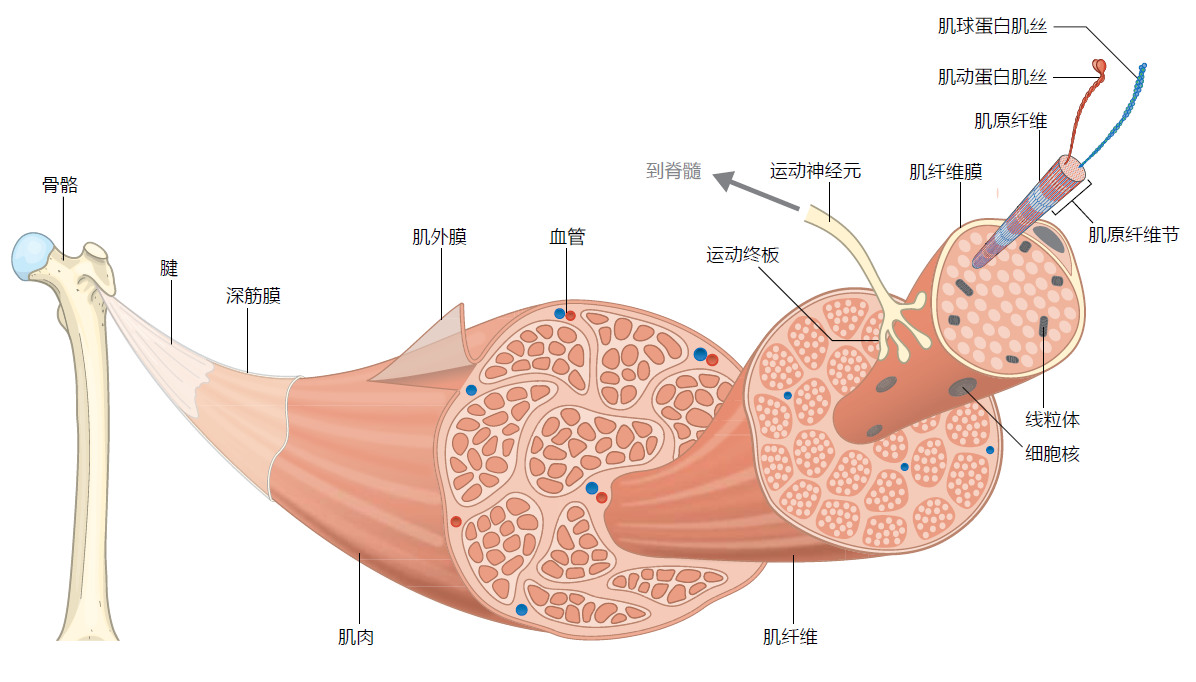
Figure 8.8 Organization of skeletal muscle. The muscles are attached to the bone via thick sheets or strands of tendon at the ends of the muscle. The body of the muscle is enveloped by the epimysium, a thin layer of connective tissue. The epimysium allows the muscle to contract while maintaining its shape. Under the epimysium lies the fascicles, bundles of muscle cells (fibers) organized into a unit by layers of connective tissue called the perimysium. The muscle fibers are long, multinucleated cells surrounded by the connective tissue endomysium. The endomysium helps to contain the capillaries and the neuromuscular junction and stem cells on each fiber. The muscle fibers contain the myofibrils, highly specialized cytoplasmic organelles containing the contractile proteins actin and myosin (see Figure 8.9). Each myofibril is surrounded by the sarcoplasmic reticulum (SR), a specialized form of the endoplasmic reticulum that stores Ca++ needed for muscle contraction (see Figure 8.13). Transverse (T) tubules connect the muscle cell's plasma membrane (also known as the sarcolemma) to the SR. The myofibrils are further organized into the sarcomere, the basic contractile unit of the muscle (seen here as the striations and in detail in Figure 8.9).
The myosin II molecule is a dimer with two globular heads and two tails coiled together extending in the opposite direction from the heads (Figure 8.9B). The two myosin heads are ATPases involved in splitting an ATP to ADP+Pi and releasing the energy needed for contraction. Approximately 200–300 individual myosin II molecules bind by their tails to form the myosin filament (Figure 8.9C). Tail-to-tail binding of the myosin II molecules gives the myosin filament a bidirectional orientation, each side of the myosin II bipolar head points away from the middle of the filament. The myosin molecule is composed of heavy and light chains. The heavy chains have a molecular weight of about 200 kd and make up the majority of the myosin head and all of the tail. The light chains have molecular weights of 20 kd and are found in the “neck” region of the molecule connecting the myosin head to the tail. Different isoforms of the myosin heavy chain (MHC) determine the contractile speed of the sarcomere.
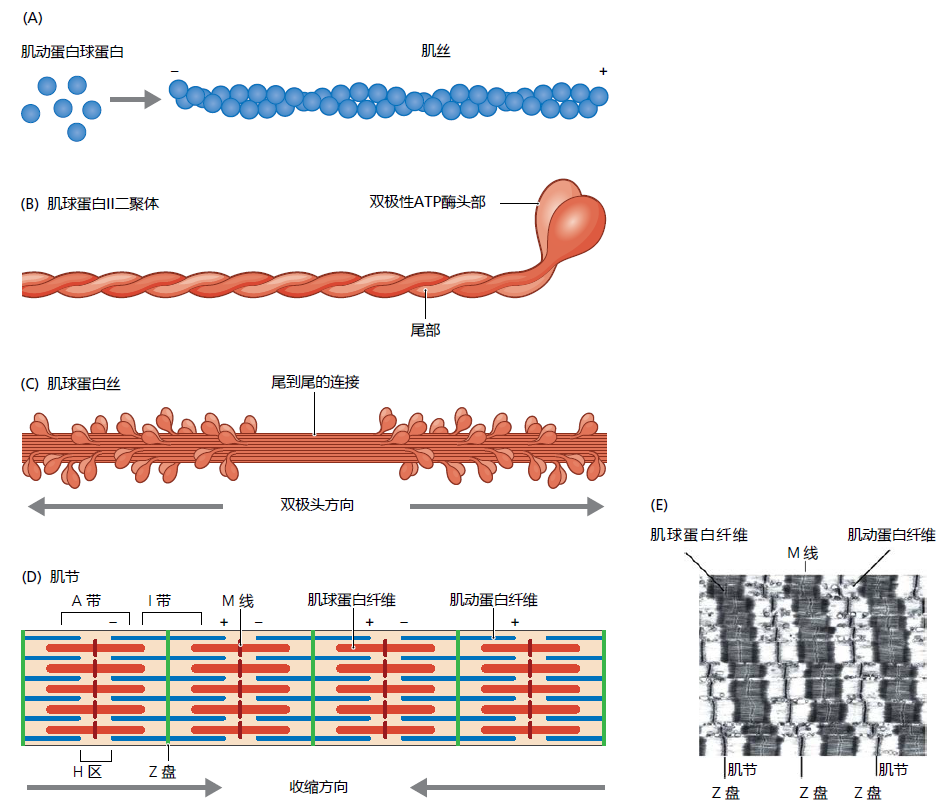
Figure 8.9 The organization of the actin and myosin filaments within the sarcomere. (A) The actin globular proteins form a polar coiled filament that provides gaps or clefts allowing for the binding of ATP and other contraction regulatory proteins. (B) Myosin II dimer showing the coiled structure of the tails. (C) Tailto-tail binding ensures that the bipolar ATPase heads face in opposite directions from each other. (D, E) The juxtaposition of the actin and myosin filament creates the basic structure of the sarcomere (E: longitudinal electron micrograph of a myofibril). (Adapted from Alberts B et al. 2014. Essential Cell Biology. New York, NY: Garland Science, Figures 17.38 and 17.40. With permission.)
The actin and myosin filaments are organized in a crossover pattern that makes up the basic contractile unit of the muscle, the sarcomere (Figure 8.9D and E). Muscle contraction occurs when millions of microscopic sarcomeres contained in the myofibrils shorten simultaneously. Shortening of the sarcomere is a cycle that begins when the myosin head, without a bound ATP molecule, attaches to the actin filament (Figure 8.10). When the muscle receives the neurologic signal to contract (see text that follows), molecules of ATP attach to the heads on the myosin filament. The binding of ATP causes a conformational change in the myosin head that, in turn, results in two concurrent actions: (1) the myosin head releases from the actin filament and (2) the myosin heads pivot about 4–6 µm in the direction of the actin's positive pole (think of the movement something like cocking the hammer on a gun). Then, the myosin ATPase hydrolyses the ATP to ATP to ADP + P i causing the opposite conformational changes to the myosin head. That is, the hydrolysis of ATP causes the myosin head to first reattach to the actin filament and then pivot back to its original position. Because the pivot occurs after the reattachment of the myosin head to the actin filament, the two filaments slide past each other. This brings the Z-discs closer together, shortening the sarcomere.
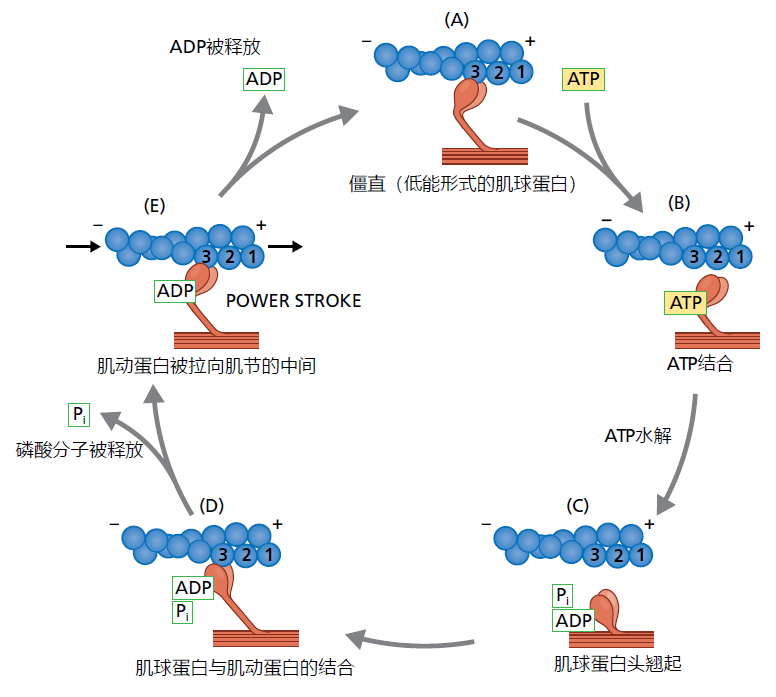
Figure 8.10 Actin/myosin molecular cycle that causes shortening of the sarcomere. (A) The cellular signal to contract causes the myosin head without ATP to bind at position two of the actin filament. (B) The myosin head releases from the actin upon binding of the ATP and “cocks” toward the positive pole of the actin molecule. (C) The hydrolysis of ATP causes the re-binding of the myosin head at position three. (D) Disassociation of the phosphate group causes the myosin head to return to its original position while bound to position three on the actin; the actin filament “slides” past the myosin filament toward the negative pole. Because actin is attached to the Z-disc at the positive pole, the sarcomere shortens. (E) The myosin head disassociates from the actin and the sarcomere relaxes, returning to its original length.
A myosin head without an ATP or ADP molecule will remain in contact with the actin filament, a state known as rigor (in death, rigor mortis). In Figure 8.10A it is shown as such only for ease of presentation. Under normal physiological conditions and without receiving a signal to contract, an ATP molecule will almost always be attached to the myosin head. The ATP-bound myosin head reflects the true resting state of the muscle, when the sarcomeres are at their greatest length. This biochemical arrangement allows for the almost instantaneous shortening of the sarcomere once a signal to contract has been received. The shortening cycle, from ATP hydrolysis to rebinding of ATP (Figure 8.10B and C), occurs in about 0.01 second.
Process of skeletal muscle contraction begins as neurologic signal
The series of events resulting in muscle contraction originates in the spinal cord where an electrical signal is transmitted to the muscle via motor neurons (Figure 8.11). One motor neuron branches and makes contact with multiple muscle fibers at the neuromuscular junction (NMJ), the motor endplate. Each muscle fiber has one, and only one, NMJ. The motor neuron, the motor endplate, and the muscle fibers innervated by that motor neuron's axonal terminals are collectively known as the motor unit. The number of muscle fibers that are innervated by a single motor neuron depends on the type of movement the muscle performs. Where fine motor control is required, such as in the hands and fingers, the motor neuron will innervate only a small number of fibers (0–200). Motor neurons innervating muscles performing gross movements, that is, the bicep muscle on the upper arm that moves the lower arm upward, will make contact with many more muscle fibers (more than 1,000).
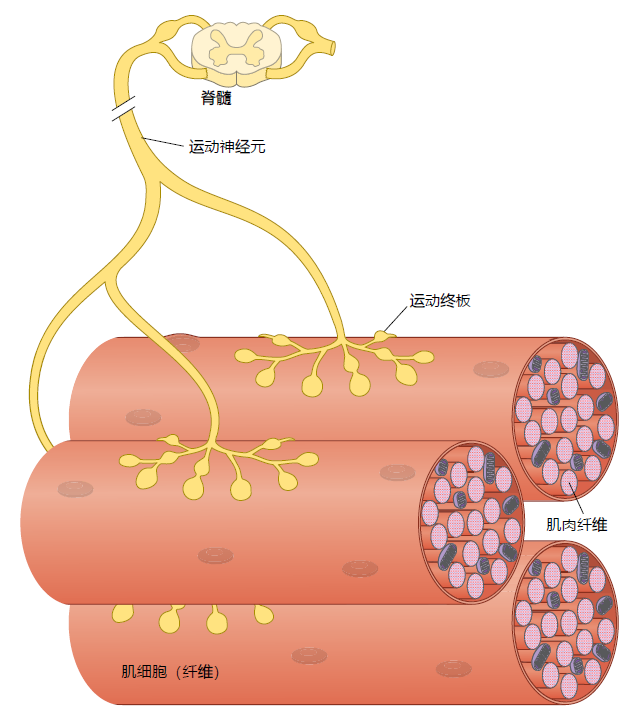
Figure 8.11 The motor unit. A motor unit consists of the motor neuron, muscle cell, and motor endplate.
Muscle contraction begins when the motor neuron, via the motor endplate, releases the neurotransmitter acetylcholine onto the muscle cell (Figure 8.12). The acetylcholine causes the plasma membrane to depolarize (reversal of the electrical charges across the cell membrane) and spread the electrical signal throughout the fiber in just a few milliseconds (the electrical signal in nerves and muscle is called an action potential and is thoroughly described in Chapter 9). The quick spread of the electrical signal ensures that all the fibers being used for contraction shorten at virtually the same time. The action potential is carried inward into the myofibril through the transverse (T) tubules, an extension of the cell membrane. The T-tubules transfer the signal to the sarcoplasmic reticulum (SR), causing Ca ++ to be released into the cytosol. Elevated calcium levels in the cytosol trigger a molecular interaction between two contraction-regulatory proteins, tropomyosin and troponin. Tropomyosin is an elongated protein that runs the length of the actin filament. When the muscle is relaxed, tropomyosin lies near the surface of the actin filament and inhibits the attachment of the myosin head to the actin filament. The binding of calcium to troponin causes a confirmation change affecting the position of the tropomyosin: the tropomyosin recedes into the interior of the actin filament. The affinity of the actin filament for myosin greatly increases, resulting in the attachment of the myosin head and progression of the contraction cycle (Figure 8.10). Termination of the neurologic signal causes Ca ++ to be sequestered back into the SR.
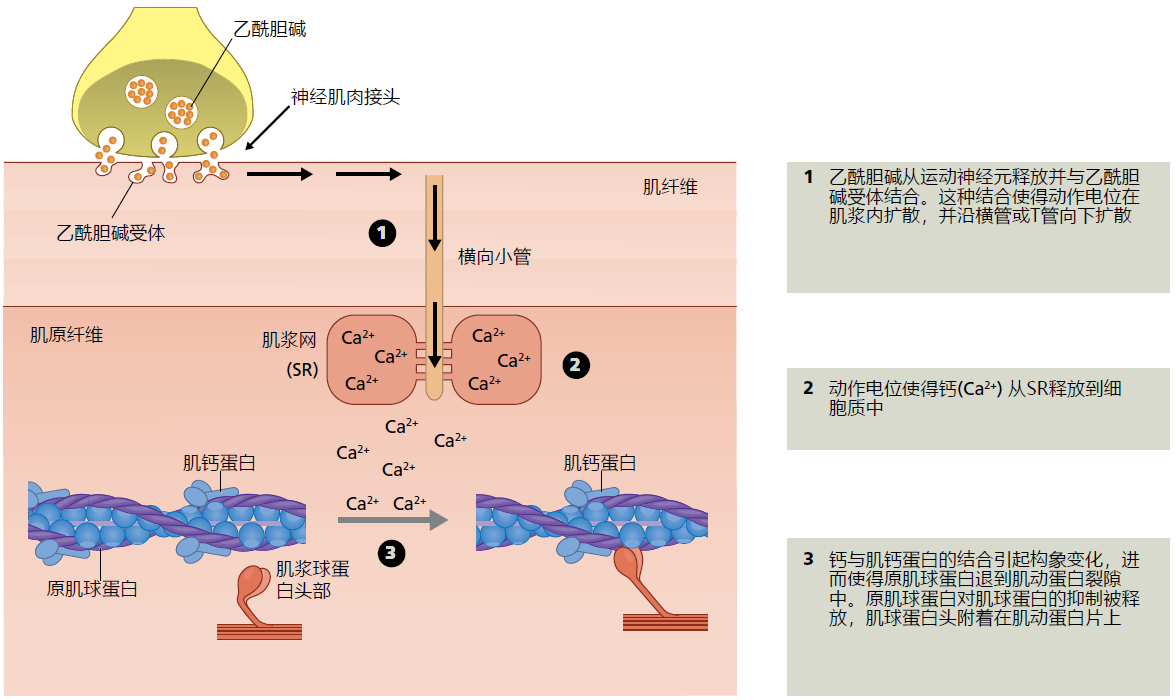
Figure 8.12 Excitation-contraction coupling. Skeletal muscle is an excitable tissue, a tissue that carries an electrical signal (action potential) along its cell membrane. In practice, excitable tissues turn an electrical signal into a biochemical or mechanical action. Here we show that the electrical signal initiated by the motor neuron and carried along the muscle's cell membrane causes the SR to release Ca2+. Calcium, in turn, interacts with troponin causing tropomyosin to release its inhibition on myosin and the start of the sarcomere shortening cycle. That is, excitation of the muscle membrane, electrical signal, is coupled with the contraction of the sarcomere, mechanical action.
Skeletal muscle contraction speed and force are determined by muscle fiber type
Recall that myosin molecules are composed of light and heavy chains, with the heavy chains having several isoforms. The myosin heavy chain (MHC) isoforms determine many of the characteristics in the muscle fiber including speed of contraction and resistance to fatigue. There are three myosin isoforms found in human skeletal muscle, type I, type IIa, and type IIx (TABLE 8.2). Type I fibers (aka, slow twitch or red) have slow contraction speed and are highly resistant to fatigue (Figure 8.13). The resistance to fatigue in type I fibers reflects their high concentration of mitochondria and reliance on oxidative metabolism (see Chapter 4, section titled, “Mitochondrial ATP synthesis produces majority of superoxide ions”). Type I fibers are important for slow, sustained locomotion, such as walking and maintaining posture. Type IIa fibers have greater contraction speed than type I fibers and are less resistant to fatigue. The decrease in resistance to fatigue in type IIa versus type I fibers results from a greater use of glycolytic metabolism (anaerobic). Type IIa fibers are likely to be recruited for faster movement such as moderate running or biking. Type IIx fibers generate the most force and power of the three types. However, type IIx fibers fatigue rather quickly because they rely almost exclusively on glycolic metabolism. They will be used for contraction when the movement requires only a brief fast and powerful contraction. The startle response is a good example where type IIx muscle will be recruited.
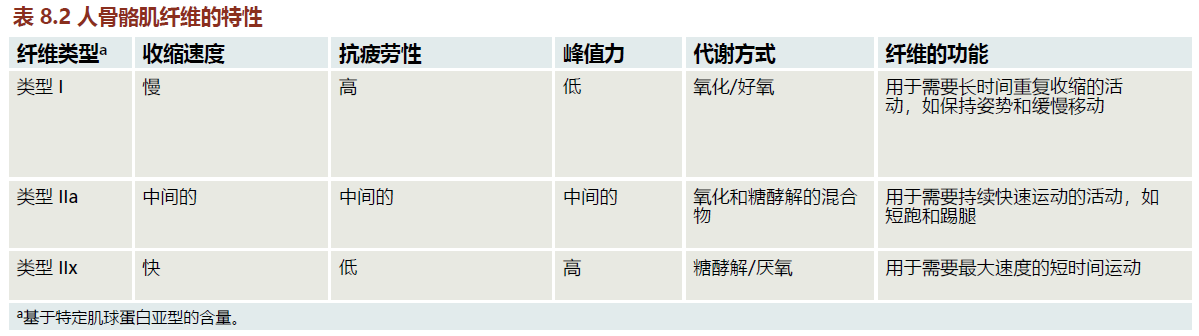
Figure 8.13 Force (Po; kilo Newtons [kN] per meter squared [m2]) versus velocity (length [L] per second [s]) for the three types of human muscle fiber. Fast, type IIx, generates the greatest force and velocity at all points of the graph compared to fast, type IIa and slow, type I muscle fibers. (From Schiaffino S, Reggiani C. 2011. Physiol Rev 91[4]:1447–1531. With permission from The American Physiological Society.)
All muscle fibers within a single motor unit are the same type, and most human skeletal muscles will contain all three types of muscle fibers. This anatomical arrangement means that a single muscle can operate at different contraction speeds depending on the movement being required. Let's use the rectus femoris muscle (RF) positioned in the front of the upper leg to demonstrate how one muscle can recruit different fibers for different movements. The RF muscle's primary function is to extend the lower leg and flex the hip—that is, bend forward from the waist. Contraction of the RF is important in stabilizing the hip and causing the lower leg to swing forward during walking and running. Type I fibers in the RF will be used for walking, because this movement requires only low-force contractions. Moreover, type I fibers are fatigue resistant, meaning that walking can be sustained over long periods. If we turn the walk into a moderate run, type IIa fibers will be recruited and provide faster contraction velocity, moving the lower leg at a quicker pace. Picking the pace up to an all-out run requires the recruitment of type IIx fibers to assist type I and type IIa fibers with fast velocity and high forces of contraction. However, type IIx fibers tire easily, and high-speed running can be maintained only for short distances.
Skeletal muscle damage repair and renewal performed by satellite cells
Satellite cells, the stem cells of the skeletal muscle, are located along the entire length of the muscle fiber (Figure 8.14). They are positioned between the sarcolemma and the basal lamina layer of the basement membrane only a few millimeters from the capillaries. The satellite cell's anatomical location within the muscle fiber assures that they are in direct contact with proteins of the extracellular matrix (ECM), the multiple layers of connective tissue that support the muscle fibers. The basal lamina of the ECM plays an important role in muscle fiber replacement and repair. Located here are proteins that secrete growth factor activating the satellite cell following muscle fiber damage. The basal lamina contains high concentrations of laminin, a glycoprotein that provides the physical space for satellite cell proliferation and migration.
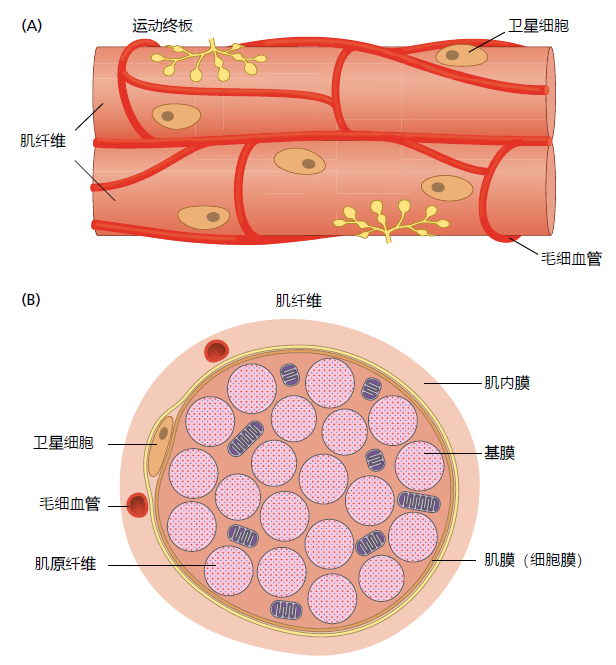
Figure 8.14 Skeletal muscle fiber, showing the anatomical locations of satellite cells. (A) Longitudinal and (B) cross-sectional views. Satellite cells are positioned between the basement membrane and the sarcolemma (cell membrane). The basement membrane, part of the extracellular matrix (ECM), is a thin sheet of connective tissue composed primarily of collagen and laminin.
The satellite cell's function in repairing and replacing damaged muscle fibers involves hundreds of regulatory factors, a description of which is well beyond the scope of this text. Here we provide only a rudimentary description of the process of muscle fiber replacement. Imagine a sprinter in a 100 m race when she or he feels intense pain in the back of the leg. The runner has just ripped apart several muscle fibers in the hamstring muscle, a pulled muscle. The damage to the muscle fibers and supporting tissues causes the release into the injury site of several proteins and other regulatory factors that otherwise would remain in the uninjured muscle fibers or the ECM. The injury also stimulates the inflammation response, an immune system process that removes the damaged tissue prior to replacement by new tissue (detailed description of the inflammation response is provided later in this chapter). Proteins released by the damaged tissue and cells of the immune systems are critical to muscle fiber replacement because they activate satellite cells.
Damage to the basal lamina results in the release of many growth factors that activate the quiescent satellite cell, the cell in G 0 (see Chapter 4, section titled, “Cell cycle and cell division”). Two growth factors found in the basal lamina that have been shown to be particular important to satellite cell activations are fibroblast growth factor 2 (FGF2) and the hepatocyte growth factor (HGF). Both FGF2 and HGF bind to specific receptors on the plasma membrane of the satellite cell. Activation of the quiescent satellite cell can also be caused by elements associated with the inflammatory response. For example, nitric oxide (NO), a free radical that acts as a biomessenger, can be found almost instantaneously after injury to a muscle fiber. Nitric oxide has been shown to induce the release of FGF2 and HGF.
Once the satellite cell has been activated, it must migrate to the site of injury prior to beginning proliferation. Migration of the satellite cell occurs through chemotaxis, the movement of cells in response to chemicals in their environment. Satellite cell migration begins when receptors on the cell membrane, known as integrins, bind to the laminin within the basal lamina. After binding to the laminin, the activated satellite cell migrates toward the damaged tissue. The activated satellite cell finds the damaged area by following the concentration gradient established by the chemotactic factors released by the ECM, injured cells, and the immune cells (Figure 8.15). That is, the satellite cell bound to the laminin senses the increasingly higher concentration of chemotactic factors as it nears the damage and moves in that direction. In general, the chemotactic factors directing the satellite cell to the area of damage are the same growth factors and inflammation response proteins that activate the satellite cell (see previous discussion).
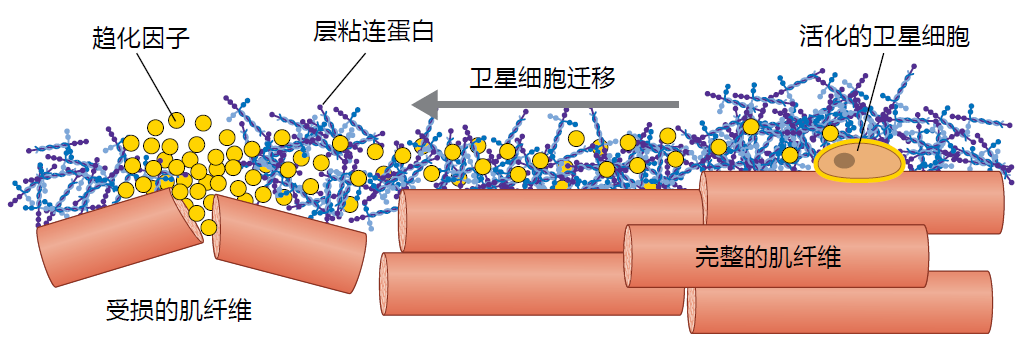
Figure 8.15 Simplified illustration of the chemotaxis mechanism in satellite cell migration. Activated satellite cells are bound to the laminin in the basal lamina. As the chemotactic factors are released by the damaged muscle fiber, they spread out across the fascicle of muscle fiber. The concentration of the chemotactic factors becomes progressively lower as it approaches the stationary activated satellite cell. The satellite cell uses this concentration gradient as a path to the damaged tissues.
Proliferation of satellite cells, that is, the reentry into the cell cycle, begins once they have reached the damaged area. The vast majority of new satellite cells will differentiate into myogenic cells, also known as myoblast, and ultimately become new or repaired muscle fibers. The skeletal muscle will also need to replenish the pool of satellite cells lost to the damage of the muscle fibers. This process is done through satellite cell self-renewal. The extracellular signals that determine whether a satellite cell differentiates into a muscle fiber or self-renews are only now beginning to be clear and involves a complex interaction of regulatory factors and genetic pathways. See the end of this chapter's “Further Reading” for more details on the satellite cell self-renewal process.
Proliferation of satellite cells destined to become muscle fibers adheres to the same general scheme as that described in Chapter 4 (section titled “Cell cycle and cell division”). After several rounds of proliferation, the myoblasts exit the cell cycle and differentiate into mononucleated myocytes. Myocytes have two fates depending on whether they are destined to become new muscle fibers or repair the existing fibers. If the myocytes receive the signal to participate in the generation of new fibers, several myocytes will fuse together and form a multinucleated structure called the myotube. Otherwise, the myocytes will fuse with the existing damaged tissue. Completion of the fusing process signals the cell to begin generating the proteins necessary for the formation of the sarcomere and other organelles of the muscle fiber.
Lack of physical activity and intrinsic aging influence time-dependent loss of muscle mass
A slow loss in muscle mass and strength occurs during the maturity phase of the human life span, a process called aging muscle atrophy. Aging muscle atrophy reflects a decrease in both muscle cell number and cell size. The loss in both cell number and size differentiates aging muscle atrophy from disuse syndromes, in which only cell size decreases. A series of cross-sectional and longitudinal studies has shown that muscle mass declines at a rate of about 8% per decade in sedentary men and 6% per decade in sedentary women (sedentary meaning that physical activity is limited to acts of daily living, cleaning the house, walking upstairs, etc.).
Investigations often cited as the bases for aging muscle atrophy have used primarily sedentary individuals. Rarely have these investigations included individuals who participate in a structured and regular physical exercise program. This seems surprising given that disuse is a major risk factor in skeletal muscle wasting. We know that older athletes have rates of skeletal muscle loss that are significantly attenuated compared to sedentary individuals of similar age (Figure 8.16). Rates of muscle mass loss in individuals who have maintained exercise are closer to 2% per decade after age 40 and do not show a gender difference like that observed in sedentary people. Additionally, aging muscle atrophy has been shown to be delayed at least a decade for individuals who exercise regularly.
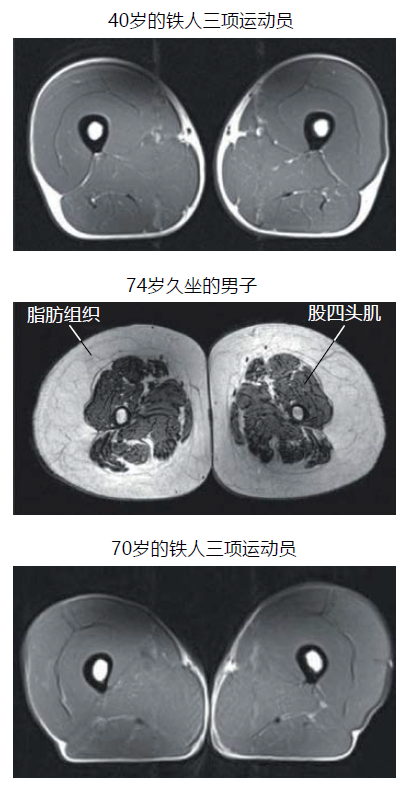
图 8.16 久坐的老年人和年轻及年长的专业 运动员大腿肌肉的磁共振成像。Individuals capable of competing in triathlons are considered elite athletes and may not be representative of a typical master athlete. Nonetheless, these images demonstrate that older individuals engaged in structured physical activity can maintain muscle mass. (From Wroblewski AP et al. 2011. Phys Sportsmed 39[3]:172–178. With permission.)
Results from studies using only sedentary individuals and those that include individuals who maintain an exercise routine provide a glimpse at the interplay between lifestyle choices and intrinsic aging. Multiple investigations have shown that lifestyle choice, choosing to exercise or not to exercise, determines the rate of aging muscle atrophy throughout most of the adult life span. Nevertheless, an intrinsic process of aging muscle atrophy will begin at some point even in the most physically fit individuals. That is, an intrinsic aging process will replace lifestyle choices as the dominant factor influencing aging muscle atrophy. At what age this switch in mechanisms, extrinsic versus intrinsic, occurs remains unknown. However, we can assume that, similar to other aging physiological declines, the timing will be highly individualized. A more thorough discussion of environmental factors that impact aging muscle atrophy, such as nutrition and exercise, is provided in Chapter 10.
Time-dependent loss in skeletal muscle strength and power correlate with aging muscle atrophy
Strength and power are often used to assess skeletal muscle functional capacity. Strength is defined as the capacity to generate maximal force at a specified velocity of muscle contraction. In practice, a velocity of zero, isometric contraction, will be used to determine strength. Since daily activities rarely require the use of maximal force or isometric contractions, many researchers and clinicians prefer to use muscle power, force times velocity, to measure functional capacity in muscle. That is, power can be measured over a wide range of velocities and forces. Muscle strength and power correlate well with the cross-sectional area (CSA) of the muscle, a measure of muscle mass. The greater is the CSA, the greater the strength and power, and vice versa. Time-dependent loss in muscle strength and power declines at about the same rate as does muscle mass, CSA, or both. The consensus from both cross-sectional and longitudinal investigations suggests that strength, power, or both, decline at a rate of 8%–10% per decade after age 40 in sedentary individuals and 2%–5% per decade in individuals who maintain an exercise routine. As would be expected, the time-dependent decrease in CSA, strength, and power in whole muscle are mirrored by similar losses in single muscle fibers (Figure 8.17).
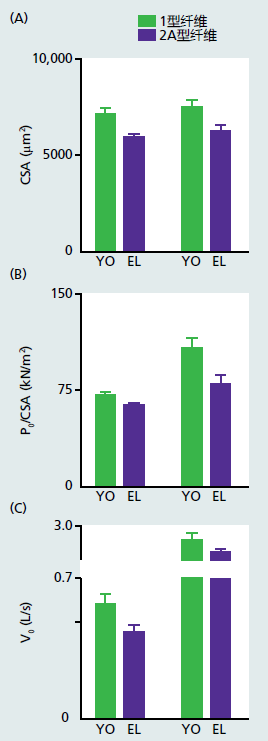
Figure 8.17 Time-dependent losses in single muscle fibers. Cross-sectional area (CSA/μm2), force (P0)/CSA and velocity (V0) in length (L) of fiber per second (s) measured in single muscle fibers isolated from young (YO) and elderly (EL) men. Both young (mean age 23.1 ± 2.2 years) and older (mean age 70.9 ± 4.4 year) were physically active. The values for all three measurements in both type I and type IIA fibers were significantly lower in the EL versus YO group. These results are consistent with other studies showing that even physically active older individuals are not immune to a time-dependent loss of skeletal muscle strength. (Adapted from Brocca L et al. 2017. J Physiol 595[14]:4823–4844, Figure 2. With permission from John Wiley and Sons.)
Recall that aging muscle atrophy can be distinguished from disuse syndromes by the loss of muscle fibers. Time-dependent loss in all three muscle fiber types has, in general, been observed in both humans and rodents. However, losses in type IIa and type IIx fibers are markedly greater than those seen in type I. These findings are consistent with the observation that muscle strength and power decline along with aging muscle atrophy. That is, type IIa and type IIx fibers generate greater force at all velocities (that is, greater power) than do type I fibers (Figure 8.13).
Intrinsic underlying mechanisms causing aging muscle atrophy are multifactorial and remain unresolved
Thousands of investigations have been completed using rodents as models to evaluate the mechanisms underlying aging muscle atrophy. Recall that investigations using rodents are carried out under strict housing and experimental conditions (see Chapter 2, Box 2.1) and remove, for the most part, environmental factors affecting the rate of aging. Rodent models used in biogerontology research are generally regarded as being most useful in describing the intrinsic process of time-dependent alterations. Therefore, our discussion here pertains primarily to intrinsic mechanisms that may lead to aging muscle atrophy.
No single mechanism or group of mechanisms associated with aging muscle atrophy has been shown to be causative; thus, the results are only observations having multiple interpretations. Many of the observations correlating aging muscle atrophy to a specific mechanism mirror the results found in other tissues as described throughout this text. For example, genetic pathways discussed in Chapter 5, such as mTOR1 and FOXO, appear to contribute to alterations in anabolism (protein synthesis) and catabolism (protein degradation) in aging muscle. A decrease in anabolism or increase in catabolism would clearly lead to aging muscle atrophy. In addition, time-dependent loss in mitochondrial function, increase in reactive oxygen species (ROS), or both, as discussed in Chapter 4, have also been linked to aging muscle atrophy. The reader should assume that the cellular, genetic, and molecular events discussed previously, and seen as general mechanisms of aging, also correlate to aging muscle atrophy. We do not further discuss those mechanisms. Here we briefly discuss two potential mechanisms that are specific to muscle and correlate well with aging muscle atrophy: alterations in neurologic innervation and function and loss of stem cell proliferation.
Denervation of motor neurons and structural fragmentation in neuromuscular junction are hallmarks of aging muscle
The motor neuron appears to undergo significant thinning, as measured by axon diameter, over time (the axon is the part of the nerve cell that makes connection to other cells; see Figure 9.2). Thinning of the axon has been shown to induce apoptosis of the nerve cell leading to complete denervation. Without the neural innervation, the muscle cells of that motor unit will atrophy, experience apoptosis, or both—that is, aging muscle atrophy. Studies in rodents suggest that there may be as much as a 20%–30% reduction of motor neuron innervation during aging. The time-dependent denervation of motor neurons occurs primarily in type IIa and type IIx motor units, an observation correlating well with loss in muscle fibers. However, whether the loss of motor neurons in aging muscle atrophy reflects the cause or the result of muscle fiber loss is far from being resolved.
The NMJ shows considerable fragmentation over time, a structural alteration that correlates with aging muscle atrophy (Figure 8.18). Whether or not the fragmentation results in decreased neurologic function remains a matter of debate. Nonetheless, the time-dependent structural alteration in the NMJ occurs to both nerve and muscle tissue, the two cell types that make up the NMJ. Electron microscopy has shown that the motor neuron and its branches at the NMJ lengthen. The lengthening of the motor neuron correlates with loss of frequency in the electrical signal arriving at the motor endplate. However, the strength of the signal, the amplitude, does not appear to change.
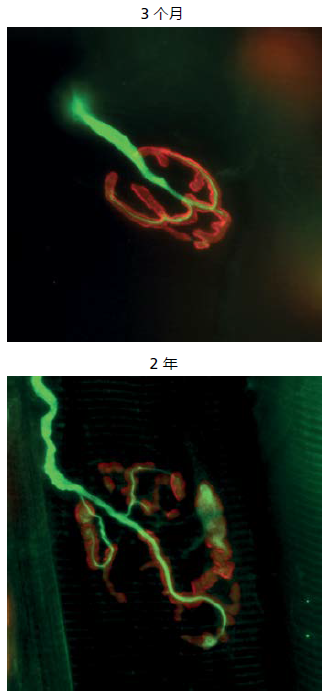
Figure 8.18 Fluorescent-labeled micrograph of a NMJ in the extensor digitorum longus muscle isolated from 3- and 24-month-old mice. Structures with green fluorescence belong to nerve tissue. Acetylcholine receptors on the membrane of the muscle fibers are visualized by the color red. Note the fragmentation of the NMJ in the 24- versus 3-month-old mouse. Fragmentation of the neuromuscular junction often leads to a disruption in the neurotransmission of the electrical signal from the nerve to the muscle. (From Tintiganc LA et al. 2015. Physiol Rev 95:809–852, Figure 7A. With permission. Image courtesy of Dr Shuo Lin.)
Fragmentation of the NMJ on the muscle membrane appears as changes to the size of the acetylcholine receptor (AChR) folds. Fusion of the folds and decreased size are most often observed. Structural changes to the AChR folds could alter the binding characteristic of the acetylcholine, although direct evidence for such changes remains to be fully resolved. A decrease in acetylcholine binding to its receptor on the muscle membrane would affect neurotransmission, the transfer of the electrical signal from the nerve to the muscle. Some, but not all, investigations have reported that neurotransmission decreases in other mammals. Observation of alterations at the NMJ remains, for the most part, at the visual level. Cellular and molecular studies are needed to understand if NMJ plays a role in aging muscle atrophy.
Satellite cell function decreases over time
Several investigations have shown that the satellite cell's ability to regenerate muscle fibers decreases with age, a finding that could explain aging muscle atrophy. Controversy exists as to whether time-dependent attenuation in the proliferative capacity of the satellite cell is a function of factors inherent to the cell (intrinsic) or factors related to the cell's local environment (extrinsic). (The term local environment refers to the blood supply, extracellular matrix, etc., that are in proximity to the satellite cell position on the muscle fiber.) Although this issue has not been resolved, current results suggest that both intrinsic and extrinsic factors impact time-dependent attenuation in satellite cell proliferation. From the cell intrinsic perspective, the pool of functional satellite cells decreases over time, suggesting that total proliferative capacity declines. Further evidence that mechanisms intrinsic to the satellite cell can contribute to diminished muscle fiber regenerative capacity comes from transplantation studies (Figure 8.19). When satellite cells are harvested from young animals and transplanted into the injured muscle of an old animal, the proliferation rate is considerably higher compared to when old satellite cells are transplanted into old muscle. However, when old satellite cells are transplanted into young animals, proliferation rates decrease as compared to normal proliferation in a young animal. If the cell's local environment, an extrinsic factor, was the only factor critical to proliferation, then the young muscle receiving the old satellite cells should not be affected.
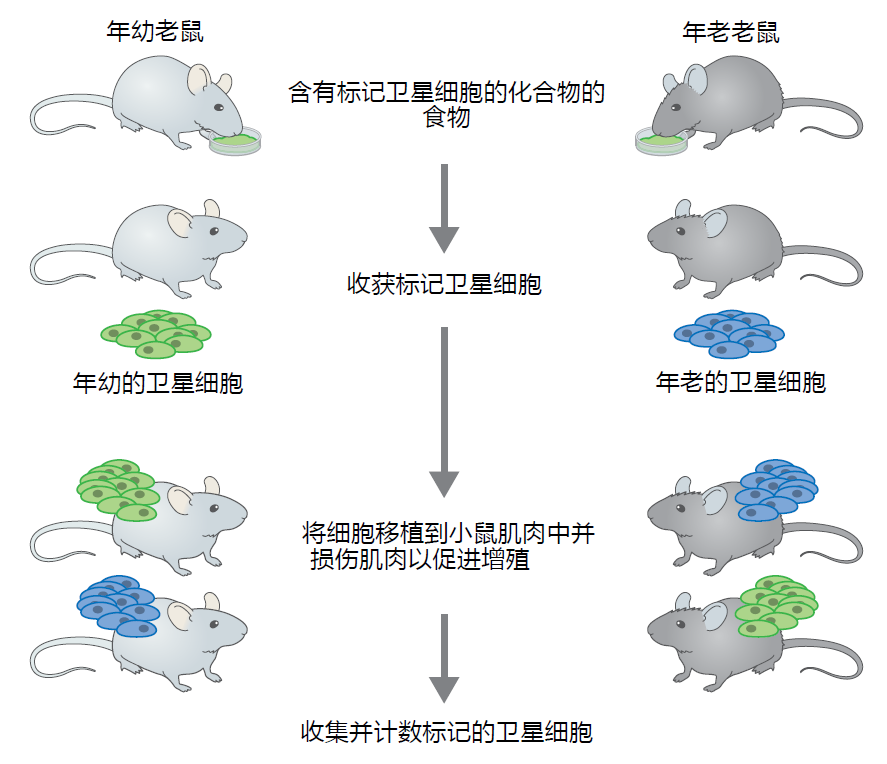
Figure 8.19 Example of an experimental design used to evaluate the effect of aging on satellite cell number and proliferation. Young and old adult mice are given a diet containing a biological compound that labels the DNA of satellite cells. Thus, the researcher has a way to follow the replication of satellite cells. After a suitable period of time, satellite cells are harvested from mice and transplanted into the muscle of differentaged mice. Young mice will receive labeled satellite cells harvested from either young or old mice. Similarly, old mice will receive satellite cells harvested from either young or old mice. By placing young cells into young mice and old cells into old mice, this study design allows the researcher to determine if the experimental procedures themselves affect the outcome (in this case, they do not). To enhance satellite cell proliferation, the muscle receiving the transplant will be injured by injection of a chemical that kills muscle fibers. Satellite cells will be harvested during the active period of proliferation and counted to determine the number of labeled cells.
Recall that the anatomical positon of the satellite cell on the muscle fiber, referred to as the niche, plays an essential role in muscle regeneration. A disruption in the architecture of the basal lamina would alter satellite cell migration to the injured site and slow the rate of muscle regeneration. Although direct evidence for time-dependent alterations in the rate of satellite cell migration is not available, it appears that the concentration of laminin in the basal membrane decreases with time. Some data suggest that the laminin is replaced with fibrous tissue. Since satellite cells must bind to laminin to migrate, these results are consistent with the hypothesis that extrinsic factors play a significant role in time-dependent alterations to satellite cell proliferation.
The most compelling evidence suggesting that extrinsic factors affect the time-dependent attenuation of satellite cell function comes from studies using parabiosis, the anatomical joining of two animals (Figure 8.20). Heterochromatic parabiosis, the joining of two animals of different ages, results in the animals sharing circulatory systems that have components of both young and old environments. Exposing the aged mouse to a younger environment results in satellite cells taking on the characteristics of a young mouse. Conversely, exposing the young animal to an old environment leads to a loss in regenerative capacity. These studies have been repeated several times and have led to the general conclusion that some factor yet to be identified in the blood can delay satellite cell aging. Research aimed at identifying the factor(s) has intensified in recent years.
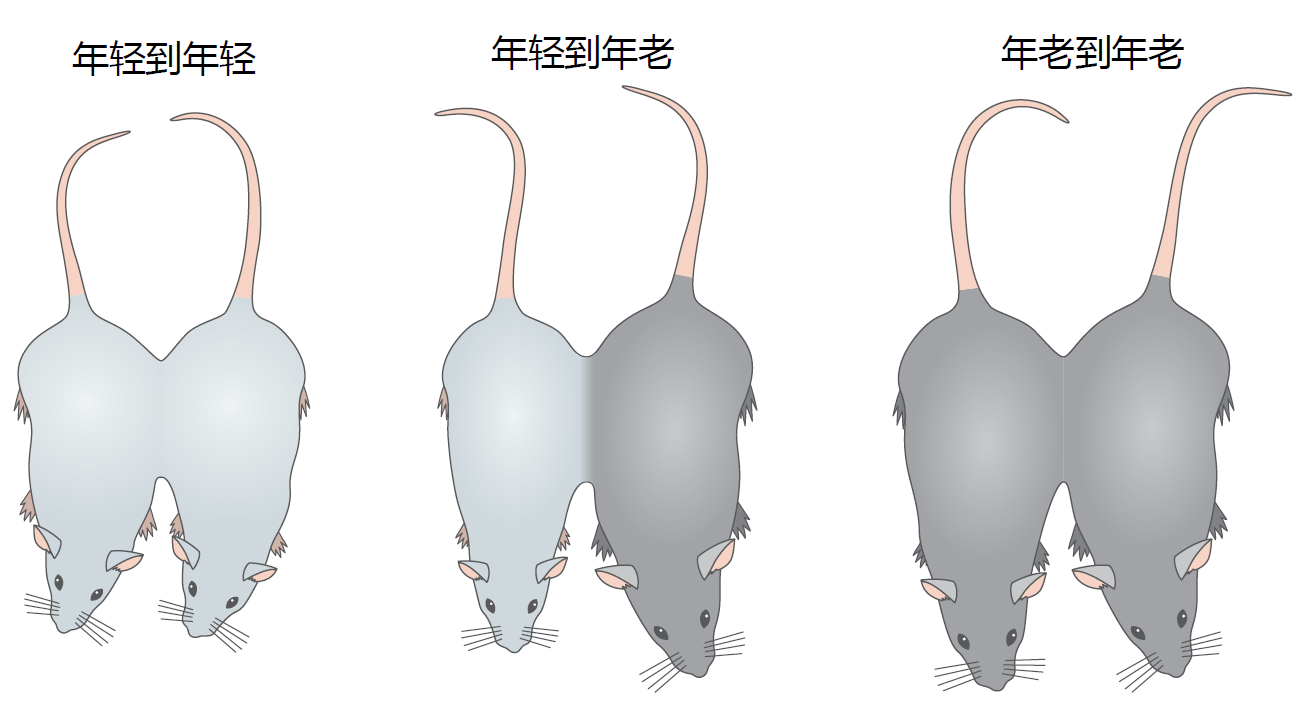
Figure 8.20 A parabiosis procedure used to determine the effect of extrinsic factors on time-dependent satellite cell function. In this example, mice are joined by suture at the knee, elbow, wall of peritoneal cavity, and skin. As the surgical injuries heal, anastomoses are formed in the circulatory system, and the two mice share blood. The sharing of the circulatory system by differentaged mice, known as heterochromatic parabiosis, exposes the young animal to an old environment and the old animal to a young environment. Chemically injuring a muscle in each mouse of the parabiotic pair and then following the rate and amount of muscle fiber regeneration allows the researcher to evaluate the effect of extrinsic regulation on satellite cell regulation.
Sarcopenia is pathological condition associated with excessive aging muscle atrophy and strength
All time-dependent skeletal muscle wasting was once defined as sarcopenia (Greek “loss of flesh”). The separation between aging muscle atrophy, a nonpathological condition, and sarcopenia, a pathological condition, was in response to the finding that a loss in muscle mass and strength in some older people correlated well with falls and loss of independence. Subsequent research showed that excessive aging muscle atrophy and loss of strength—that is, sarcopenia—were often associated with poor nutrition, isolation and depression, and chronic time-dependent disease. While sarcopenia does not have a definition or diagnostic criteria listed in the International Classification of Diseases, several working groups in North America and Europe have arrived at a consensus on a working definition: “Sarcopenia is a syndrome characterized by progressive and generalized loss of skeletal muscle mass and strength with a [increased] risk of adverse outcomes such as physical disability, poor quality of life and death.” The increased risk of disability and mortality separate the disease of sarcopenia from the normal time-dependent loss in muscle mass and strength known as aging muscle atrophy.
Estimates in the prevalence of sarcopenia vary widely, since no generally accepted diagnostic criteria have been established. Published values of prevalence in sarcopenia are as low as 14% to as high as 73% in individuals over the age of 70 years. For this reason, the U.S. National Institutes of Health and the Health Ministries in Canada and Europe have combined efforts to establish specific tests and guidelines for the diagnosis of sarcopenia. At the time of this publication, formal adoption of standard tests and diagnostic guidelines for sarcopenia has not been achieved. However, most experts in this area agree that evaluating stability, strength, and whole-body skeletal muscle mass are sufficient to diagnose sarcopenia. To this end, walking speed and grip strength are used as measures of stability and strength, respectively. Walking speed lower than 0.8 m/s and grip strength below 20 kg for women and 30 kg for men are suggestive of sarcopenia. If walking speed or grip strength are below these standards, then skeletal muscle mass standardized to the height squared should be measured using dual-energy x-ray absorptiometry. Muscle mass of <7.23 kg/m2 in men and <5.67 kg/m2 in women would indicate sarcopenia.