
5.3 在老年生物学中研究基因表达
One of the most important advances in biology occurred during the 1970s, when scientists were able to remove a piece of DNA from a chromosome and determine its sequence. The ability to extract and characterize a DNA segment led to the development of technologies that allowed scientists to manipulate a gene sequence or develop entirely new genes, then to study how these treatments affected the functions of cells and organisms. These techniques, known collectively as recombinant DNA technology, would change the direction of all research in biology. From ecology to molecular genetics, scientists could now identify specific genes and gain a more complete understanding of the fundamental mechanisms underlying the why and the how of life.
Recombinant DNA technology also spawned the boom in the commercial biotechnology industry. Techniques that were once the domain of only the most highly trained and skilled scientists were now automated and commercialized so that the nonspecialist could apply sophisticated gene analysis techniques to specific research interests. Biogerontologists began using these techniques to identify possible genes that influence the rate of aging and longevity.
You learned in Chapter 1 how biogerontologists use simple organisms such as yeast, worms, and flies to study the genetics of aging. These organisms provide the biogerontologist with several advantages, including (1) their genomes have been sequenced—that is, the sequence and locations of all genes are known; (2) the long history of general genetics research using these organisms has resulted in hundreds if not thousands of readily available mutants with a variety of genotypes and phenotypes; and (3) the life spans of these organisms are short, allowing for several longevity studies within a month (yeast) or a year (worms and flies). You also learned that any genes identified as modulating the rate of aging and longevity in these simple eukaryotes will have gene orthologs—genes having similar functions—in more complex organisms, given the phylogenetic relationships. Once a gene has been shown to affect longevity in simple eukaryotic organisms, a similar gene can be identified in a more complex organism, such as a mouse, and its possible relevance to the rate of aging or longevity in humans evaluated.
Using the enormous amount of accumulated knowledge about the genetics of these three model organisms, biogerontologists have identified a few genes that seem to directly modulate the rate of aging and longevity. The details of these discoveries are presented in the sections that follow. First, we explore the general techniques and methods used in the discovery of these longevity genes.
The general process leading to the identification of genes that modulate the rate of aging and longevity requires several steps (Figure 5.15). The complexity of this process is such that no single researcher or laboratory could possibly perform all the steps required. The gene discovery process reflects a community of researchers throughout the world, relying on one another‘s scientific ability to perform the critical steps in the process. Although the techniques and methods described briefly here are presented as a single, seamless procedure, keep in mind that each step entails, for the most part, independent laboratories completing work over several years.
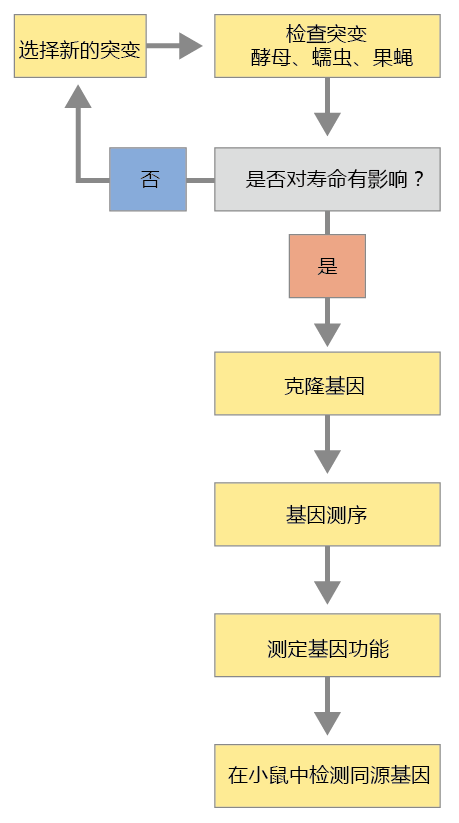
Figure 5.15 General scheme for identifying genes that affect longevity. Researchers consult computer databases or the research literature to choose a mutant strain that has a genotype or phenotype that may affect longevity. Once the mutant strain has been selected, the next step is to clone the gene. Cloning is the process of making several identical copies so as to have enough genetic material to sequence the gene. Once the gene has been sequenced, its function is determined. The final step of the process is to identify the gene's ortholog in mice and evaluate whether it has a similar effect on longevity.
5.3.1 Genetic analysis in 老年生物学 begins with the screening of mutants
By far the greatest advantage of using S. cerevisiae, C. elegans, and Drosophila for genetic analysis in aging research is that their entire genomes have been sequenced. Sequencing of a gene, however, does not provide information about the function of that gene. To gain that knowledge, geneticists start by creating mutant strains of these organisms in which a single gene has been altered to produce a specific genotype. Looking at the differences between the genotypes of the mutant strain and the wild type (that is, the organism in its normal or natural condition, as distinct from an atypical or mutant type) gives scientists the first information about the function of that gene. A biogerontologist with a particular hypothesis about how genes affect the rate of aging and longevity can then select mutant strains showing genotypes consistent with a particular hypothesis. (Because most research is performed with public money, researchers are obligated to make their mutant strain available to other scientists.) In general, biogerontologists select strains with mutations affecting growth and/or reproduction. (Recall, from Chapter 3, the close relationship between growth, reproduction, and longevity.) Since many genes affect growth and reproduction, the biogerontologist must select several different mutants and then compare their longevity characteristics to determine which one best suits his or her needs.
The comparing of mutant strains, known as genetic screening, may seem relatively easy and straightforward. However, these beginning steps of genetic analysis are the most critical and the most difficult. Most genes are pleiotropic (a single gene producing more than one phenotype), and mutant strains often show several phenotypes associated with the mutation of a single gene. Even if one of the mutant strains has a desirable longevity characteristic, it may also have a phenotype that could confound life span and cause significant variation in results. Thus, the genetic biogerontologist may have to refine the mutation so that only the longevity phenotype is present. Refining the genome so that there is only one genotype or phenotype resulting from one gene mutation requires meticulous research, with lots of trial and error that ends in failure. It can take years for a researcher to succeed in developing a mutant that provides a suitable and reliable experimental system for testing a hypothesis about the impact of genes on longevity.
5.3.2 Identification of gene function requires DNA cloning
Even after a scientist has developed a reliable mutant strain with a phenotype having extended longevity, the function and regulation of the gene, as well as the structure of the 蛋白质 it encodes, will be unknown. To determine the function of a gene, it must be cloned—that is, identical copies of the gene must be created. By cloning a gene, the researcher produces enough DNA material to determine the gene's base-pair sequence. Knowing the base-pair sequence, the researcher can determine the amino acid sequence of the 蛋白质 it encodes and thus the 蛋白质‘s possible function.
The cloning process was once a very laborious task requiring several different procedures, perhaps taking days to complete. Today, these procedures are automated and combined into a single process known as polymerase chain reaction (PCR), which takes only hours to complete (Figure 5.16). The process begins with the isolation of either DNA or mRNA from tissue or cells, procedures that are fairly easy. DNA is used as the starting sequence if the scientist needs to clone the entire genomic sequence of the gene, since genomic DNA includes both exons (the mRNA-coding sequences) and introns. If determining the amino acid sequence of a 蛋白质 is the objective, only the exon sequence of the gene is needed, and the starting material is mRNA (recall that mRNA is the complementary product of only the exons). Here we describe the process using mRNA as the starting material.
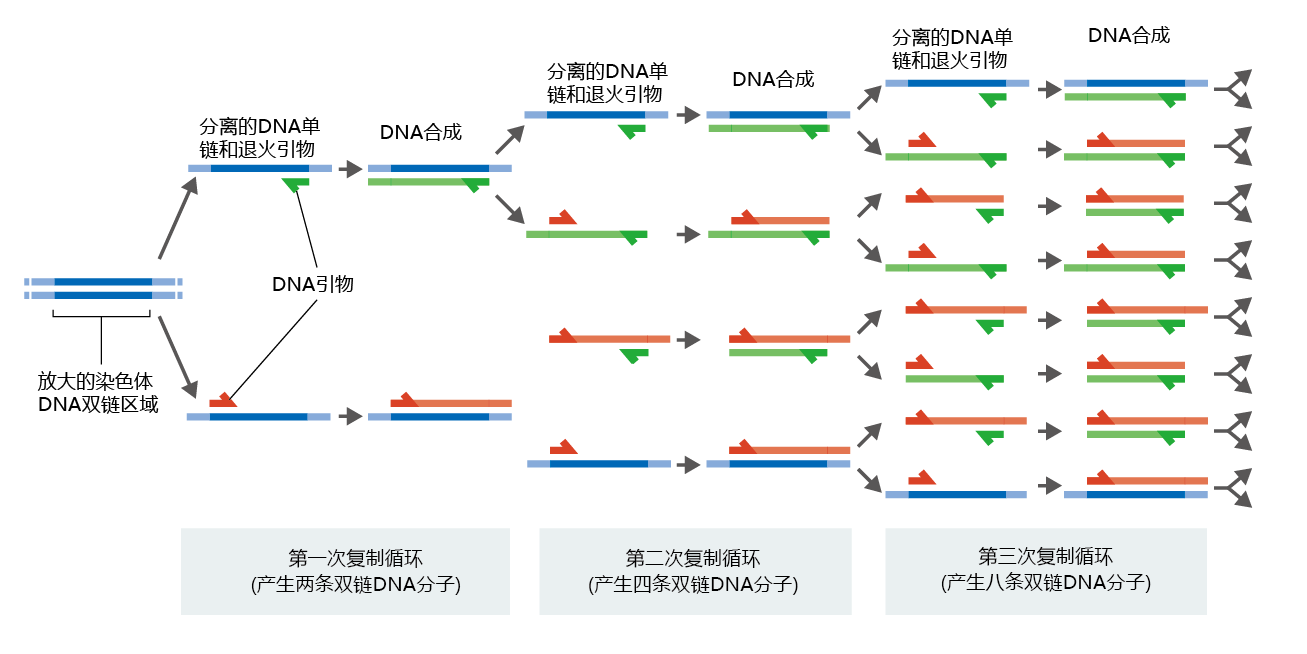
Figure 5.16 Cloning a gene using PCR. After identification of the segment of DNA to be cloned, the DNA is heated to separate the strands, and short complementary sequences known as primers are attached to the 3′ end of the sequence of interest. The primers direct a reverse transcriptase to the start of the gene. The DNA is then mixed with a DNA polymerase and deoxyribonucleoside triphosphates, and the DNA is replicated to create two daughter DNA molecules. Repeating the process multiple times, using the newly synthesized fragments as templates, quickly amplifies the number of copies.
Before beginning the PCR process, the researcher must design and chemically create a set of oligonucleotides (oligo- meaning “many”) that are exclusively complementary to the 3′ end of the gene being isolated. The 3′ end of the gene is known because the sequence of the gene being used to develop the mutant is known. In everyday practice, the researcher designs the oligonucleotides and sends those plans to a company for synthesis. When the oligonucleotides, or “primers,” are added to a solution containing the mRNA, they direct an enzyme called a reverse transcriptase (which makes DNA from mRNA, the reverse of the usual process in cells) to start replicating the gene being amplified. The deoxyribonucleotides necessary to make DNA are also added to the solution. The reverse transcriptase uses the mRNA template to make a new, complementary DNA (cDNA) strand containing only the exons from the gene being isolated.
At this point in the process, the mRNA and the new cDNA are bound together by hydrogen bonds in a double strand and must be separated so that only the cDNA is amplified. The PCR machine heats the solution to around 90°C to break the hydrogen bonds. DNA polymerase and a second set of primers, complementary to the 3′ end of the cDNA, are then added, and a new strand of cDNA complementary to the first cDNA is made. The PCR machine slowly cools the solution, and the two complementary strands of cDNA form a double helix. The amplification process is now a simple matter of heating and cooling the solution in the presence of deoxyribonucleotides and DNA polymerase. Each cycle doubles the number of identical genes. It takes about 30 cycles to produce enough material for sequencing the gene.
5.3.3 The function of the gene can be partially determined from its sequence
Like PCR, gene sequencing is now automated. The newly synthesized gene copies are added to a solution that breaks the cDNA into fragments terminating at specific bases. Because there are literally billions of copies of the new gene, cDNA fragments are generated that terminate at every position on the gene. The robotics of the sequencing machine mix reagents that add a fluorescent dye to the terminal base of the cDNA fragment. Each base is labeled with a different color. The fragments are separated by atomic weight, using gel electrophoresis (Figure 5.17), which is also automated by the sequencing machine. Based on the presence of the fluorescent dyes, detectors in the sequencing machine read the position of each fragment on the gel relative to the other fragments, and with the aid of computers, the sequencer determines the sequence of the gene.
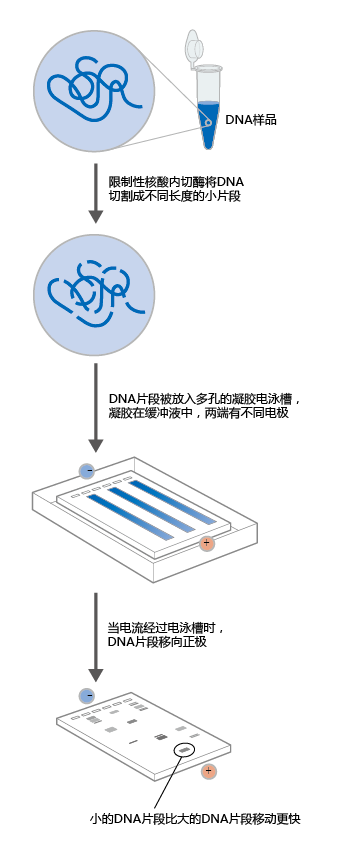
Figure 5.17 Gel electrophoresis. Electrophoresis on a porous gel physically separates DNA molecules on the basis of their size and electrical charge.
The final step in this process involves applying the genetic code (see Table 5.1) to the gene sequence to determine the amino acid sequence of the 蛋白质 it encodes and thus its probable function. However, the raw gene sequence does not tell us where the first set of three bases that codes for a specific amino acid (that is, the first codon) starts. At this stage, the researcher can only make an educated prediction about the function of the gene, based on the collective knowledge of previous work in genetics. The National Institutes of Health maintains a large computer database of gene sequences with known functions. (Once a gene‘s function is determined, researchers submit their gene sequence to this database.) Computer software has been developed that allows researchers to compare their sequence to sequences of known genes and receive information on the probable function of their gene. Researchers can use this information to focus their research and, using recombinant technology, can determine more precisely certain characteristics of their gene, including the start and stop codons.
5.3.4 In situ hybridization can reveal a gene‘s function
Determining the function of a gene often starts with an evaluation of where and when the gene is expressed. To accomplish this, a technique call in situ hybridization (in situ is Latin for “in place”) can be used. Since the sequence of the gene is known, segments of the gene can be labeled with a detectable and measurable compound, such as a radioactive isotope or fluorescent dye, and introduced into cells, tissues, or whole organisms. When these labeled nucleic acid probes encounter a complementary sequence of mRNA, they bind to that sequence and create a hybrid molecule. The hybrid molecules can then be isolated and measured using the appropriate detection machines.
In situ hybridization is one more technique that can be used to evaluate the expression pattern of a gene. For example, suppose we determined, using computer databases, the most likely function of a gene extracted from C. elegans. Our analysis suggests that the gene‘s sequence has high homology with genes that express their mRNA in response to certain ambient temperatures. Because our gene is unique, we don't know what that temperature might be. We can determine the temperature, and thus the function of the gene, by injecting C. elegans with labeled DNA probes and then expose the animals to different temperatures. The greatest in situ hybridization will occur when a temperature is reached that causes the greatest gene expression. In addition, this experiment provides information about which cells or tissues have the greatest expression of the gene. As you‘ll see later in the chapter, an experiment similar in design to this hypothetical one was used to determine which cells in C. elegans express a 蛋白质 associated with enhanced longevity.
5.3.5 Genetically altering organisms helps evaluate a gene's impact on human 寿命
As you'll recall, one of the many reasons that a genetic biogerontologist begins genetic analysis with simple organisms is that they have shorter life spans than more complex organisms. This means that scientists can screen for longevity genes in a relatively short amount of time. Once the gene has been cloned and sequenced, and its function determined in simple organisms, mutant strains of more complex animals with physiology closer to that of humans can be developed. In general, the mouse has been the mammal of choice for genetic analysis of the rate of aging and longevity.
A common method for evaluating a gene‘s influence on the rate of aging or longevity is to develop mutant strains of mice that either have an extra copy of the gene or do not have the gene at all. Comparing these mutants with the wild-type mouse demonstrates whether the gene alters the rate of aging or longevity. Having determined which mouse gene to modify, it is necessary to know whether gene expression or gene silencing caused the increase in longevity, knowledge that is gained from analysis of the simpler organisms. If repression (gene silencing) causes the longevity effect, then the gene ortholog is removed from the mouse‘s genome. This type of mutant is called a gene knockout. If expression of the gene causes an increase in longevity, adding an extra gene results in greater expression than is observed in the wild-type mouse. This type of mutant is called a transgenic organism. Figure 5.18 illustrates how transgenic and knockout genes are produced for use in mice. Once one of these genes has been created, it is amplified and inserted into mouse embryos and will then be incorporated into the mouse‘s genome.
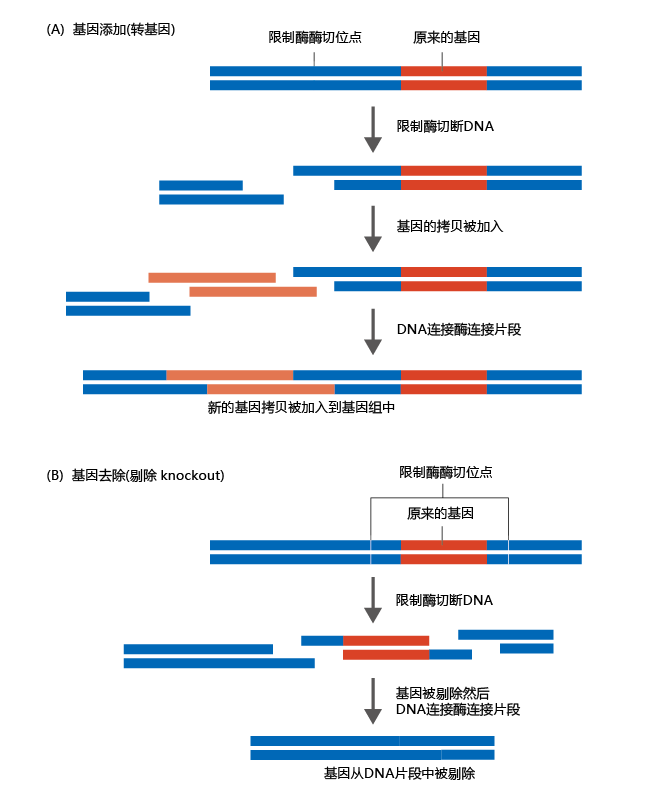
Figure 5.18 Making a transgenic or knockout gene for use in mice. (A) Gene addition. After cloning the gene ortholog, a fragment of the DNA with the original gene is exposed to restriction nucleases. The restriction enzymes cleave the DNA at specific sequences. The cloned gene is then added to a mixture containing DNA ligase, an enzyme that stitches DNA together at specific sites. The final product is a DNA fragment that has two copies of the same gene. (B) Gene removal. Restriction enzymes and DNA ligase are used to remove the identified gene, leaving the mouse genome without the gene of interest (such as a suspected longevity gene).
5.3.6 DNA microarrays are used to evaluate 基因表达 patterns at different ages
Our discussion to this point has focused on recombinant DNA technology used to determine a single gene‘s function(s). However, in the real world, a cell may express hundreds and maybe thousands of genes in response to a single stimulus or cellular event. Until the mid-1990s, determining the gene expression pattern in response to a single stimulus was impossible. The advent of hybridization techniques led to the development of a tool called DNA microarrays, which allow researchers to determine the gene expression pattern of a cell at a single point in time.
DNA microarrays are pieces of glass, about the size of a postage stamp, that have been embedded with thousands of single-stranded DNA sequences from specific genes with known or unknown functions. Robots are used to precisely place the DNA sequences on the glass so that their position is known. Then, mRNA is extracted from the cell of interest and converted to cDNA, which is labeled with fluorescent probes. The microarray and the cDNA probes are incubated together, allowing hybridization to occur. The microarrays are then washed to remove unbound probes, and the hybridized molecules are identified by their fluorescence, using automated microscopes. Figure 5.19 shows this process, using a hypothetical example of analyzing mRNA isolated from the muscle cells of mice at two different ages.
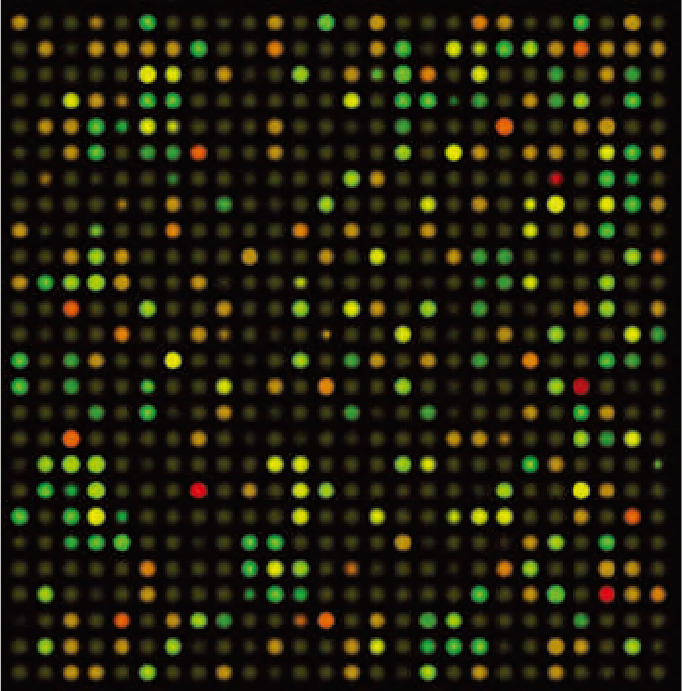
Figure 5.19 DNA microarrays reveal 基因表达 patterns. DNA microarrays are often used to evaluate 基因表达 patterns in two samples treated with a different stimulus or obtained under different conditions. In this hypothetical example, mRNA was isolated from muscle cells of mice at two different ages. After converting the mRNA into cDNA, the fragments are labeled with a red (young mice) or a green (old mice) fluorescent dye and mixed together. The solution is allowed to hybridize to the cDNA fragments on the microarray. After the unbound fragments are washed off, the fluorescence is scanned by an automated detector. Green dots indicate greater gene expression in the old mice; red dots, greater expression in the young mice; yellow dots, expression in both young and old mice; and brown dots, no expression of the gene by either group. (Courtesy of Alila Medical Images/Shutterstock.)
Experiments similar to the hypothetical example shown in Figure 5.19 have been carried out. The results were mixed. Differences in gene expression between young (3–6 months) and old (26–30 months) mice were extremely modest and depended on the organ from which the mRNA had been isolated. For none of the organs did the difference between young and old mice exceed 3% of the total genes represented on the microarray, with most organs showing only about a 1.0–1.5% difference. These values are largely considered statistically insignificant. Moreover, the gene expression pattern of the old mice was not consistent across organs. That is, the age-related gene expression pattern in the heart was not the same as that observed in the brain, which was not the same as that in the muscle, and so on. These results are consistent with the proposition that aging is not a result of a genetic program but more closely reflects random stochastic events that vary considerably among tissue types.
The results of investigations designed to evaluate age-related gene expression patterns suggest that the usefulness of microarray technology to biogerontology may be in determining how interventions that delay aging alter the gene expression pattern. For example, we now know that maintaining a healthy weight by reducing caloric intake can significantly slow the rate of aging of many organ systems in humans. The knowledge that this effect may be directly related to an age-related alteration in the gene expression pattern arises from microarray studies performed using cDNA from mice undergoing calorie restriction. Restricting the caloric intake to 70–80% of calories normally consumed by mice given free access to food results in a significant reduction in functional loss in all physiological systems (this is further described in Chapter 10). Comparison of gene expression profiles between free eating and calorie-restricted aged mice shows dramatic differences. Although differences in the gene expression profile vary from organ to organ, and how these differences directly affect the rate of aging has yet to be determined, these results provide solid evidence that genes can affect the rate of aging and the life span. The identification of specific genes that respond to interventions will greatly advance our knowledge of the factors contributing to inherited differences in the rate of aging.